Translating timepoints from mouse trials into human clinical trials
A new compound, anle138b, has recently been touted as the most promising anti-prion drug candidate yet discovered [Wagner 2013 (ft, supp1, supp2)]. And rightly so: it is orally bioavailable, crosses the blood-brain barrier, shows no toxicity in animals even at enormous doses, and has fairly large effects on survival even when animals are treated late in the course of prion disease.
That word – ‘late’ – is very important here. Hosts of compounds have shown an ability to delay symptoms of prion disease if administered early in the incubation period, long before symptoms. But 85% of prion disease cases in humans are sporadic CJD patients [Appleby & Lyketsos 2011], who will never know they have a disease until they are already sick. A compound that is going to work for these people has to work in mice that are already showing symptoms of disease.
But that turns out to be a tricky thing to quantify, because whether mice are showing symptoms depends in part on how hard you look. Mallucci’s first paper on Cre-mediated PrP knockout in mice [Mallucci 2003] didn’t report any symptoms prior to 10 weeks post infection (wpi), but a few years later, through more meticulous behavioral testing, she was able to find certain symptoms starting at 8 wpi, in the exact same mouse model [Mallucci 2007].
Wagner 2013 makes the claim that the mice treated with anle138b at 80 dpi and 120 dpi were treated “after onset of disease”. That is certainly true for 120 dpi – Wagner provides his own data for weight loss beginning by that time – but for 80 dpi he cites Mallucci 2007. That’s where comparisons get tricky. Mallucci found behavioral symptoms at 8 wpi = 56 dpi, in a model where mice die at ~12 wpi = 84 dpi. That’s a much faster mouse model than Wagner’s model, in which controls live to ~180 dpi, so symptoms may well appear earlier.
That prompts me to re-iterate my statement from the anle138b post: different prion mouse models get sick at different times. Incubation time of prions in mice is exquisitely correlated with both the PrP expression level of the mouse and the infectious titer of the inoculum. It’s also affected by prion strain and mouse background. So comparisons between models need to adjust accordingly.
Whether or not Wagner’s mice had symptoms at 80 dpi, it looks like they definitely did by 120 dpi. Yet other investigators have treated similar mouse models at similar timepoints – say, treatment at 100 dpi in a model where controls lived to 180 dpi in [Riemer 2008 (ft)] – and not made the claim of treating already-sick mice. That’s probably just because, again, finding early symptoms depends on how hard you look. Importantly, Wagner’s mice were not at the stage that researchers call ‘clinical signs’ or ‘clinical onset’ (impaired gait, etc) at the time when they began treatment.
Terms like ‘late’ and ‘after onset’ are a bit subjective, and to anchor this conversation in real past experience, I think a comparison to pentosan polysulfate (PPS) is merited. Wagner et al largely omit PPS from their summary of other proposed anti-prion compounds (Fig S20 on p. 31 of supp1), because they only consider compounds that have been administered peripherally to animals infected intracerebrally. I completely agree on the importance of only considering such stringent mouse models going forward, but by omitting the original PPS experiments in mice [Doh-Ura 2004], Wagner et al have left out an important piece of prion history.
Doh-Ura used a very rapid mouse model in which controls die at 51 dpi. Doh-Ura didn’t look hard for early symptoms; he reported that the animals exhibit “ambiguous signs of reduced activity about 2 days prior to death”. Because PPS does not cross the blood-brain barrier, it was administered by continuous intraventricular infusion with a surgically implanted cannula. It extended survival by a huge amount in mice infected with 263K prions (the primary model used), but its effects were time-dependent. It more than doubled incubation time if administered starting at 7 dpi, but had no significant effect when given at 42 dpi. Because the raw numbers in terms of days post-infection are so different between Doh-Ura’s study and Wagner’s study, I find it useful to convert dpi numbers into a new figure as follows:
%diseasecourse = treatment started dpi / control survival
And I also convert therapeutic effects into a percentage delay:
%delay = treatment survival / control survival – 1
When I do this and compare PPS with anle138b, I get the following table (PPS numbers are eyeballed from Doh-Ura Fig 2B since he doesn’t give the raw data):
| drug | treatment started dpi | treatment survival | control survival | %delay | %diseasecourse |
|---|---|---|---|---|---|
| PPS 460ug/kg/day | 7 | 125 | 51 | 145% | 14% |
| PPS 460ug/kg/day | 21 | 95 | 51 | 86% | 41% |
| PPS 460ug/kg/day | 35 | 75 | 51 | 47% | 69% |
| PPS 460ug/kg/day | 42 | 51 | 51 | 0% | 82% |
| anle138b 5mg/day | 0 | 355 | 180 | 97% | 0% |
| anle138b 5mg/day | 80 | 242 | 168 | 44% | 48% |
| anle138b 5mg/day | 120 | 224 | 172 | 30% | 70% |
You can see that Doh-Ura’s trial of PPS at 42 dpi was ~80% of the way through the disease course, later than any timepoint tested by Wagner. The fact that Doh-Ura reported no symptoms until ‘ambiguous signs of reduced activity’ at 49 dpi while Wagner reported that 80 and 120 dpi (respectively 48% and 70% of the way through the control survival time) already constituted ‘treatment after onset of disease’ might reflect real differences in mouse models, but more likely just reflects how hard they looked for symptoms.
When I plot these data in R:
ppsx=c(14,41,69,82)
ppsy=c(145,86,47,0)
anlex=c(0,48,70)
anley=c(97,44,30)
main = "extension of survival of PPS vs. anle138b"
sub = "PPS [Doh-Ura 2004], anle138b [Wagner 2013]"
xlab = "% of disease course that had passed before treatment began"
ylab = "% extension of survival in treatment group vs. control"
plot(ppsx,ppsy,col="purple",pch=19,xlim=c(-.01,100),xlab=xlab,ylab=ylab,main=main,sub=sub)
points(anlex,anley,col="orange",pch=19)
points(anlex,anley,col="orange",type="l")
points(ppsx,ppsy,col="purple",type="l")
legend("topright",legend=c("PPS","anle138b"),col=c("purple","orange"),pch=19)
Then you can see the difference in survival between the two compounds:
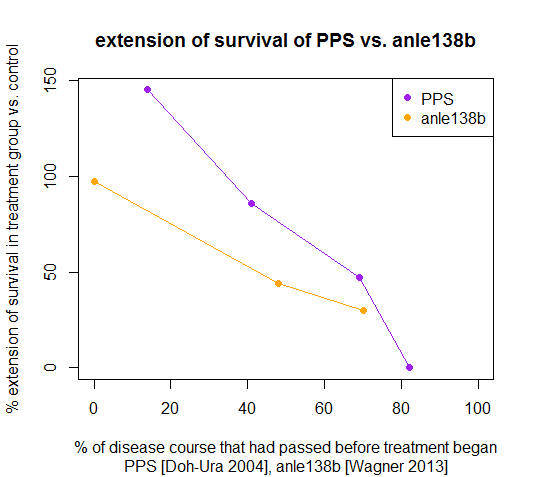
By these measures, PPS was actually more effective at all tested timepoints in terms of extending survival than anle138b, and yet still it had no effect when it was administered at 42 dpi, ~80% of the way through the disease course, a later timepoint than any tested by Wagner.
In spite of the evidence that PPS did not work late in disease, it went on to clinical trials in Japan and the U.K., which were roundly unsuccessful. To be sure, PPS was a rather problematic drug which had to be infused intraventricularly via cannula in humans just as it had been in mice. But PPS didn’t fail clinical trials due to cannula-related complications (though there were some of those too), it failed on efficacy, showing no effect at all on survival of treated patients.
Maybe that was due to strain specificity or other problems in translating drugs from mouse to humans, but one can make a good argument that the timepoint of treatment is sufficient to explain PPS’s failure. If you read carefully the official report on the U.K. clinical trial [Bone 2008], you’ll see that most of the patients were already unable to walk by the time they began pentosan polysulfate treatment. That’s a lot worse than Doh-Ura’s “ambiguous signs of reduced activity” which the mice reached only in their last day or two of life, and probably represents a timepoint far later (perhaps at ~170-180 dpi) than any timepoint where Wagner began treating mice with anle-138b.
True, you can notice symptoms in mice very early if you look hard enough. And one would certainly hope that we humans are better at recognizing symptoms in our fellow humans than we are at recognizing symptoms in mice. It could be speculated that some even earlier timepoint in Wagner’s mice – 30, 40, 50 dpi? – corresponds to when we would notice symptoms in humans.
However, the path from first symptoms to a diagnosis can be long, at least when there is no family history of prion disease. From the time when patients first show symptoms, family members need to notice, become worried, see a doctor, get referred to a specialist, and then the specialist has to run a battery of tests and puzzle over what the problem is. sCJD is notorious for appearing to neurologists as lyme disease, a viral infection, an adverse drug reaction, or a host of other possible conditions [Murray 2011]. Once a diagnosis finally is made, then also allow time for a referral to a specialized center such as Cleveland Clinic or MRC Prion Unit. No wonder the patients treated with PPS were in such bad shape by the time they finally entered the clinical trial.
So treating mice at a time when some subtle behavioral symptoms or weight loss are present may not represent a timepoint at which humans will be treated. On the other hand, if we were to simply accept that patients will not be available for treatment until they are already in such a debilitated state, then we are basically accepting that no treatment is possible. By the time a patient is bedridden, a large fraction of neurons in the most-affected brain regions have probably already been lost, and though a total knockout of PrP (were it possible) would still stop the spread of prions, it would not bring back cognitive function, because neurogenesis in adults is fairly limited.
Therefore we simply need to find a way to act earlier. Mallucci has made an excellent case for the existence of a window for intervention in prion disease where neurons are impaired but not yet lost [White & Mallucci 2009, Moreno 2012]. Mallucci’s stage is one where some behavioral symptoms are present in mice and so it may correspond to a stage in humans where we could diagnose the disease, even if we aren’t doing so consistently yet.
And having a treatment available would itself drive earlier diagnosis. Murray’s diagnostic paper, oriented towards clinical neurologists, is very enlightening in this regard. The abstract declares that the paper “highlights the relatively rare treatable mimics which must not be missed” (my underlining). Table 2 lists about 15 other conditions (some as broad as ‘viral encephalitis’) that can be difficult to distinguish from sCJD, and – this is crucial – the caption on the table reads “Those in bold represent potentially treatable conditions which must not be missed” (again, my underlining). The paper discusses the various diagnostic tools for sCJD including the test for 14-3-3 protein but finally, in the Practice Points, it summarizes that, for sCJD, “Disease duration is the most reliable distinguishing feature” [Murray 2011].
This is the practice of neurology today: there are a lot of conditions that can cause overlapping sets of symptoms, and it is difficult to wade through them all to find the right diagnosis, so doctors justifiably prioritize looking at the diagnoses that would be treatable. So simply having a treatment available will make a difference in diagnosis – it will get CJD to be one of the bold items in Murray’s list.
Despite the critical view I’ve taken here of how ‘late’ 80 dpi and 120 dpi really are, I actually do not disagree at all with Wagner’s general claim that anle138b is the most promising treatment yet proposed for prion diseases. Its BBB permeability, oral bioavailability, low toxicity, large and (by in vitro PMCA at least) non-strain-specific inhibitory effects are a compelling package of attributes that no previous drug has combined. I am hopeful that it will make its way to clinical trials soon. When it does, it will take an all-out effort to get patients treated as early as humanly possible.
And, when it does, we will also need to consider the ambiguous questions about what ‘success’ will look like. From what we see in mice, anle138b does not reverse disease, it just extends survival. Until we have a clinical trial, we are unlikley to know whether or not that will translate into any improvement in quality of life for patients who are already debilitated by prion diseases. If a drug extends life without improvement in quality of life, can we call that a success? Clinical trials follow pre-defined clinical endpoints, of which time to death is often one, so yes, a drug could potentially be approved even if it didn’t improve quality of life. If such a drug got approved, would that be a success?
I tend to think yes, because the existence of an approved drug would further raise public and physician awareness and the priority placed on diagnoses of prion disease, making it possible for patients to be treated earlier and earlier in the disease course when it might start to make a real quality of life difference too. And it would also be our first step at fighting these diseases, to be combined with future treatments yet to be discovered.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.