Structural mutants of PrP
Post last updated September 1, 2014
Over 30 different structurally mutated versions of PrP have been observed naturally or experimentally. Understanding the behavior of these mutant PrPs can help to teach us what parts of PrP are required for prion replication and for neurotoxicity. The mutants which were studied in mice and in N2a cells up to 2008 were reviewed elegantly in Aguzzi 2008. This post is intended to provide an encyclopedic reference to all these mutants, including those observed in humans and other species. Please comment if anything is incorrect or missing. My aim will be to keep this post up to date with all the mutants reported in the literature as time goes on and provide a detailed description of each.
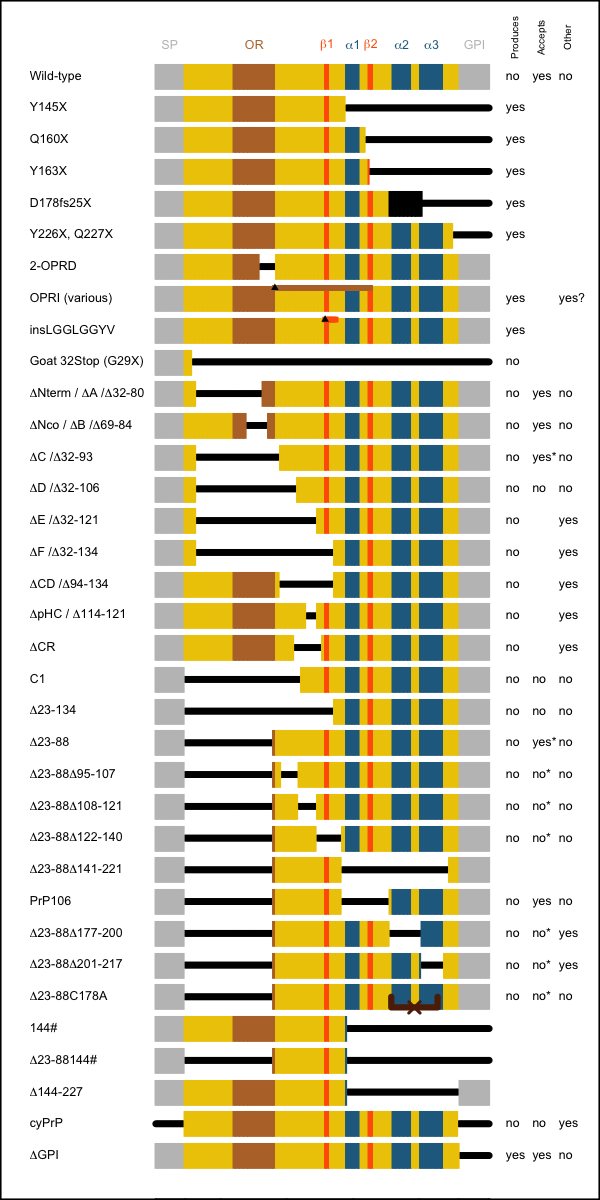
This table summarizes all of the reported structural mutants. Detailed descriptions follow. The columns at right indicate:
- Produces prions – does this PrP sequence, expressed in vivo, spontaneously give rise to prions? For the purposes of this column I’ll accept amyloid formation as evidence of a prion having been formed – but see sections below for more details on transmissibility to mice.
- Accepts prions – does this PrP sequence, expressed in vivo, support prion replication – i.e. can humans with this allele develop prion disease, or can this allele’s protein product be converted to PrPSc in the mouse brain? Note that several sequences have been described to not support prion replication in ScN2a cells, but that I do not consider this to evidence against replication competence, since some PrP mutants only support prion disease in vivo after a very long incubation time, at least on first passage, and it is not clear that N2a cells provide the amount of opportunity for prion adaptation that is afforded in the living brain.
- Other phenotype – does this PrP sequence, expressed in vivo, give rise to any other spontaneous phenotype?
I have left these columns blank where the answer has not been characterized.
Code that created this graphic
HuPrP Y145X

This truncating mutation has been observed in one patient with cerebral amyloid angiopathy [Kitamoto 1993, Ghetti 1996]. It has never been transmitted to mice, but the PrP amyloid plaques in this disease do include the wild-type allele’s protein product, as evidenced by the immunoreactivity of plaques to antisera against PrP220-231, a region not present in the Y145X allele [Ghetti 1996]. To me this seems to be evidence of intermolecular transmissibility at a minimum, even if interorganismal transmissibility has not been observed. Attempts to model this mutation in the mouse have been unsuccessful (see 144# below).
HuPrP Q160X

This truncating mutation has been reported a few times [Finckh 2000a, Finckh 2000b] but was described in the greatest detail by [Jayadev 2011] in a proband and her mother. One of the two had chronic diarrhea, but unlike in the Y163X pedigree (see below) this occurred only after onset of dementia. The PrP plaques did not react with C-terminal antibodies, suggesting no involvement of the wild-type allele. Transmissibility was not tested.
HuPrP Y163X

Of the nonsense mutations observed in humans, Y163X is the only one that has been extensively characterized and observed segregating with disease in a family [Mead 2013]. It leads to eventually to cerebral amyloid angiopathy, but first has a decades-long disease course with peripheral PrP plaque formation, chronic diarrhea and polyneuropathy. Transmission to mice was unsuccessful, but as with Y145X, the plaques reacted with a C-terminal antibody (Pri-917, against amino acids 216–221), suggesting that the wild-type allele had been recruited. The Y163X mutation was first reported in [Revesz 2009].
HuPrP D178fs25X

This 2-bp frameshift deletion at codon 178 creates a secreted mutant PrP with a cerebral amyloid angiopathy phenotype apparently similar to the nonsense mutations above and below, and diarrhea, perhaps reminiscent of Y163X. This was only characterized in one patient, but family history indicated a parent and grandparent with the same condition [Matsuzono 2013]. Transmissibility was not tested.
HuPrP Y226X & Q227X

I’m lumping these two together since they are at adjacent codons and leave virtually the entire protein intact – in fact, the stop codons are only one or two residues away from the site where ADAM10 cleaves PrP in its natural proteolytic shedding process. These have each been observed in one patient with cerebral amyloid angiopathy, and transmissibility has not been tested [Jansen 2010].
Goat 32Stop
This naturally occurring nonsense mutation, homologous to G29X in HuPrP numbering, was observed in a Norwegian dairy goat herd [Benestad 2012]. It would leave only a 7 amino acid peptide after signal peptide cleavage, and appears to be effectively a knockout allele. Neither heterozygotes nor homozygotes were reported to have any phenotype.
HuPrP OPRD mutants

A single octapeptide repeat deletion (1-OPRD) is a neutral variant found in Mediterranean populations [Palmer 1993], with about a ~1% allele frequency. 2-OPRD alleles have been reported in two patients with prion disease [Beck 2001, Capellari 2002]. Transmissibility was not tested.
HuPrP OPRI mutants / MoPrP PG14

There is a vast literature on octapeptide repeat insertions (OPRIs) in humans, which I’m not terribly familiar with and deserves a separate post at some point. OPRIs of varying lengths have been reported. 1-, 2- and 3-OPRI have only been in single case reports and may not be pathogenic, 4-OPRI has been seen more often but may be incompletely penetrant [Kaski 2011] while ≥ 5-OPRI can be somewhat more confidently characterized as Mendelian disease alleles [Mead 2007, Mead 2006, Kumar 2011]. The OPRI mutations have a fairly early age of onset (e.g. 35 for 6-OPRI), a long disease course (several years), and are transmissible to HuPrP mice [Mead 2006].
Prior to the report of 12-OPRI [Kumar 2011], the largest known mutation was 9-OPRI, known as PG14 (9 inserted repeats plus 4 endogenous repeats plus 1 nonarepeat = 14, and the repeats are rich in proline and glycine, hence PG14). A mouse model of this PG14 mutation [Chiesa 1998] developed spontaneous neurological illness characterized by cerebellar granule cell loss, even with only about 1x expression of the mutant protein. The disease was fastest on a Prnp knockout background but was still present on a wild-type background. The PrP in the brains of these mice exhibited some properties of PrPSc including detergent insolubility and some (limited) degree of protease resistance, but the disease could not be transmitted to wild-type mice [Chiesa 2003].
HuPrP 8-amino acid insertion (insLGGLGGYV)

This in-frame 24 base pair duplication event was reported, apparently de novo, in a single patient with GSS [Hinnell 2011]. The authors simply refer to it as “a novel prion protein gene mutation” or as “365-388 dup”; here I’ve named it after the eight amino acids which were inserted after codon 129. The patient had an early onset (34 years old) of an atypical dementia, but seemed to have been healthy prior to onset. Transmissibility was not tested.
MoPrP ΔNterm / ΔA / Δ32-80

Known by various names, this was the first mutant expressed in mice [Fischer 1996]. Because PrPSc can have its N terminus digested by proteinase K or removed by beta cleavage and yet retain its infectivity, this experiment was designed to test the hypothesis that PrP without the N terminus could still support prion disease in vivo. The extreme N-terminal tip of the finished peptide, amino acids 23-31 — KKRPKPGGW — were left in simply because the authors weren’t sure whether PrP would be properly directed to the ER and have its signal peptide cleaved without them. It turns out (see Δ23-88 below) that PrP does localize properly without those nine amino acids. The ΔNterm mice had no spontaneous phenotype, but could be infected with RML prions and had pathology and incubation times similar to mice expressing wild-type PrP.
MoPrP ΔNco / ΔB / Δ69-84

Also reported in [Fischer 1996]. Similar to ΔNterm, this mutant had no spontaneous phenotype but seemed to support prion disease upon inoculation just like wild-type PrP.
MoPrP ΔC / Δ32-93

These mice were characterized in Shmerling 1998 as having no spontaneous phenotype, and their susceptibility to prions was examined in Flechsig 2000. The mice were susceptible to RML prions, but only weakly so. Their incubation times were longer than wild-type mice, even at high levels of transgene expression (e.g. ~260 days to terminal illness at a 4x expression level), they produced prion infectivity titers that were 10-30x lower than wild-type mice and NaPTA-precipitable PrPSc levels that were 50x lower, and they exhibited neuropathology only in the spinal cord. This reduced susceptibility to RML didn’t appear to be due to a transmission barrier, because incubation times were not reduced on second passage. There was some ambiguity here, though – the second passage mice did have some (albeit limited) pathology in the brain, unlike the first passage mice.
MoPrP ΔD / Δ32-106

These mice were characterized in Shmerling 1998 as having no spontaneous phenotype, but their susceptibility to prions was never formally reported. However, both Weissman & Flechsig 2003 and Aguzzi 2008 cite “unpublished data” to state that these mice were not susceptible to prions.
MoPrP ΔE / Δ32-121

Shmerling 1998 reported that these mice have a spontaneous fatal disease involving neuronal loss in the cerebellum. The disease is most pronounced when the mutant allele is expressed on a PrP knockout background; it can be dose-dependently rescued by wild-type PrP. They were unable to transmit this disease to Tga20 mice, which overexpress wild-type PrP.
MoPrP ΔF / Δ32-134

Shmerling 1998 reported that these mice the same spontaneous cerebellar disease as Δ23-121 mice, but with a slightly later onset. As with Δ23-121, disease is most pronounced when the mutant allele is expressed on a PrP knockout background; it can be dose-dependently rescued (i.e. onset delayed) by wild-type PrP. They were unable to transmit this disease to Tga20 mice, which overexpress wild-type PrP.
The Harris lab also now uses the Δ23-134 construct and has shown that, like ΔCR (below), it makes cells hypersensitive to cationic antibotics [Massignan 2010].
MoPrP ΔCD / Δ94-134

Looking at the phenotypes of the A – F mice, Aguzzi observed that it was only the deletions (E and F) that extended into the “Central Domain” (CD) of the protein that caused spontaneous disease. The ΔCD mice were therefore created to test the hypothesis that only the central domain had to be deleted in order to cause spontaneous cerebellar disease. Sure enough, the ΔCD mice got a disease like that of ΔE and ΔF mice, but with an even earlier onset and a requirement of more wild-type PrP to rescue it [Baumann 2007].
MoPrP ΔpHC / Δ114-121

The “deletion of part of the Hydrophobic Core” or ΔpHC mice were created along with the ΔCD mice to see whether a smaller deletion would cause the same phenotype [Baumann 2007]. The ΔpHC mice had no spontaneous phenotype, yet the mutant did behave differently than wild-type PrP. PrPΔpHC was less good than wild-type PrP at rescuing the ΔF phenotype, giving only a marginal (p = .06) delay in disease onset, and it actually made the ΔCD disease even more rapid. The authors concluded that ΔpHC is a “partial agonist.” See more discussion at the bottom of this post.
MoPrP ΔCR / Δ105-125

This creation of the Harris lab is even more lethal than ΔCD, which was the most lethal of the Zurich series of mutants. Δ105-125, in which the highly conserved Central Region is deleted, makes cells hypersensitive to cationic antibotics such as zeocin [Massignan 2010], causes spontaneous transmembrane currents [Solomon 2010], and, when expressed on a knockout background, causes lethal cerebellar granule cell loss within 1 week after birth, even at 1x expression levels or less [Li 2007]. Co-expression of wild-type PrP can rescue this phenotype, though it takes 5x wild-type PrP to extend life to about 1 year. The phenotype of these mice seems almost identical to that of ΔCD mice; the age of onset is, if anything, even earlier and the amount of wild-type PrP required for rescue even greater.
C1 / MoPrP Δ23-111

As introduced here, PrP undergoes a proteolytic event called alpha cleavage which produces the N1 and C1 fragments. It has recently been shown that there are at least three different alpha cleavage sites [McDonald 2014], but the first identified site was on either side of H111 (human sequence) / H110 (mouse sequence) [Harris 1993]. The Harris lab therefore created a mouse which constitutively expresses only the C1 fragment – in fact, they deleted up through 111 (mouse numbering), leaving a protein which begins with the AGAAAAGA palindromic region [Westergard 2011]. The mice had no spontaneous phenotype, and could not be infected with prions. In fact, co-expression of C1 with wild-type PrP extended the incubation time for RML prions, implying that the C1 fragment is actually dominant negative against prion propagation.
MoPrP Δ23-134

These mice were also created by the Harris lab and, like the C1 mice, don’t develop any spontaneous phenotype (even with higher expression levels than the Δ32-134 mice above, which Harris used as foils to these mice) and are not susceptible to prion infection [Westergard 2011]. Like C1, this fragment appears to be dominant negative against scrapie infection – when co-expressed with wild-type PrP, it prolongs the incubation period, though the effect is smaller than with C1.
MHM2 and MoPrP Δ23-88

MHM2 is a chimeric molecule composed of mouse PrP with 2 hamsterized amino acids (109 and 112 in hamster/human numbering) completing the 3F4 epitope (KTNMKHM, aa 106-112 in hamster/human numbering). The MHM2 Δ23-88 mutant additionally has much of the N terminus lopped off, including the N-terminal tip. The Prusiner lab originally expressed this mutant in N2a cells and showed that it was capable of converting to PK-resistant PrPSc [Rogers 1993], then expressed it in mice and showed that it caused no spontaneous phenotype [Muramoto 1997]. The next question was whether these mice were susceptible to prion disease, and the answer turned out to be complicated. Mice expressing only MHM2 Δ23-88 could not be infected with RML prions at all [Supattapone 1999]. Co-expression of one allele of wild-type MoPrP, however, rendered these mice susceptible [Supattapone 1999]. It wasn’t just that only the wild-type allele was supporting the prion replication, however, because (1) the incubation time was 257 days, compared to > 400 days for mice expressing only one wild-type allele, and moreover, (2) some of the PK-resistant PrPSc reacted with the 3F4 antibody, which recognizes MHM2 Δ23-88 and not wild-type MoPrP.
A couple years later, they did a more detailed characterization to unravel why the MHM2 Δ23-88 sequence was so resistant to mouse prions [Supattapone 2001]. In this study, they compared mice with and without the 3F4 epitope and the 23-88 deletion: MoPrP, MHM2, MoPrP Δ23-88, and MHM2 Δ23-88. They found that no matter what they did, they simply could not transmit prions to mice that only expressed MHM2 Δ23-88. Yet surprisingly, mice expressing only MoPrP Δ23-88 were susceptible to RML prions, as were mice that expressed MHM2 without the deletion. The eventual disease phenotype and neuropathology of the MoPrP Δ23-88 mice were described as representing “typical signs of scrapie”, yet the incubation time was surprisingly long: 160 days on first passage and 140 days on second passage, compared to ~50 days in Tg4053 mice overexpressing wild-type MoPrP, even though the MoPrP Δ23-88 mice expressed twice as much PrP as Tg4053 mice did. Mice expressing full-length MHM2 PrP had similarly extended incubation times. Therefore the authors concluded that the resistance of MHM2 Δ23-88 mice to prion infection was the result of an interaction between the fundamentally lower susceptibility of MHM2 PrP to prions and the fundamentally lower susceptibility of Δ23-88 PrP to prions.
The crucial piece of this experiment that is missing for me is that they never tested the prion infectivity titers in the brains of the MoPrP Δ23-88 mice at the time of terminal disease. Hypothesis: if residues 23-88 are involved in a crucial neurotoxic pathway, as much evidence supports [Sonati 2013], then the long incubation times might be due to reduced neurotoxicity, not to reduced prion replication. If so, then by the time they finally do succumb to prion disease, they should have had time to accumulate much greater infectivity in their brains than mice expressing wild-type PrP.
MHM2 Δ23-88 Δ95-107 / ΔSTE

This and the next several mutants were created by the Prusiner lab in a systematic campaign to figure out which regions of PrP secondary structure were required for prion replication. The 95-107 segment (96-108 in human numbering) deleted in this particular mutant was considered to be the “stop transfer effector” (STE) because its series of positive charges (HSQWNKPSKPKTN in HuPrP) was predicted to help direct insertion of the hydrophobic core into the membrane in transmembrane forms of PrP – see this post for some cell biology basics.
In ScN2a cells, MHM2 Δ23-88 supported prion replication (though in the presence of wild-type PrP) [Rogers 1993], but additionally deleting 95-107 rendered the molecule unable to convert to PrPSc in ScN2a cells [Muramoto 1996]. When expressed in transgenic mice on a knockout background, MHM2 Δ23-88 Δ95-107 caused no spontaneous phenotype [Muramoto 1997]. The susceptibility of these mice to prions was never tested.
MHM2 Δ23-88 Δ108-121

When the Prusiner lab went on its campaign of deleting PrP secondary structure regions, it used a computational prediction of the structure of PrPC which later turned out not to be entirely accurate. MoPrP residues 108-121 (109-122 in human numbering), deleted in this mutant, were predicted to be an alpha helix dubbed H1, but the first NMR structure of the globular domain of PrPC, published the same year [Riek 1996], showed that there was in fact no alpha helix here. In any event, this mutant did not support prion replication in ScN2a cells [Muramoto 1996] and caused no phenotype in transgenic mice [Muramoto 1997]. The susceptibility of the mice to prions was never studied.
MHM2 Δ23-88 Δ122-140

This mutant did not support prion replication in ScN2a cells [Muramoto 1996]. Alone among the Prusiner series of MHM2 mutants, this one was never successfully expressed in transgenic mice. It is not known whether this is due to bad luck, toxicity or something else.
MHM2 Δ23-88 Δ141-221

Possibly apocryphal. Aguzzi 2008 refers to such a mutant, citing [Muramoto 1997], yet as far as I can tell this mutant is not present in the Muramoto paper.
MHM2 Δ23-88 Δ141-176 / PrP106 / “miniprions”

Alone among the Prusiner series of mutants, MHM2 Δ23-88 Δ141-176 supported prion replication in cell culture [Muramoto 1996]. Residues 141-176 (human numbering 142-177) comprise alpha helix 1, beta sheet 2, and the loop between beta sheet 2 and alpha helix 2. Because these deletions leave only a 106-residue, GPI-anchored peptide, the protein was dubbed PrP106. There was no spontaneous phenotype in vivo [Muramoto 1997].
The susceptibility of these mice to prion infection was extensively studied [Supattapone 1999] and was even referenced in Prusiner’s Nobel lecture [Prusiner 1998]. Tg(PrP106) mice were susceptible to RML prions in 300 days on first passage, and 66 days on second passage. It was proposed that the “miniprions” created in these mice might be useful for structural studies of PrP, but if these studies were done I never saw the paper on them. The neuropathology in the mice was described as “characteristic of experimental scrapie”.
MHM2 Δ23-88 Δ177-200

According to Riek 1996, α-helix 2 encompasses amino acids 179-193 (mouse numbering). This Prusiner lab deletion mutant therefore includes all of &alpha-helix 2 and a bit extra on both sides, though in the papers [Muramoto 1996, Muramoto 1997] it is incorrectly referred to as H3. This molecule couldn’t be converted to PrPSc in ScN2a cells, so the susceptibility of mice was never tested. The male mice expressing this mutant did, however, suffer a fatal neuronal storage disease, with cytoplasmic neuronal inclusions of insoluble mutant PrP [Muramoto 1997]. The phenotype looked quite different from that of scrapie infection, and the transmissibility of this disease was never tested.
MHM2 Δ23-88 Δ201-217

Similar to the above, this was the Prusiner lab’s deletion of α-helix 3 (which spans 201-218 [Riek 1996]), which they originally called H4. Like Δ23-88 Δ177-200, this mutant did not convert to PrPSc in ScN2a cells [Muramoto 1996], but caused a spontaneous neuronal storage disease in mice [Muramoto 1997]. This deletion was more fully penetrant than the Δ177-200 deletion and affected both male and female mice.
MHM2 Δ23-88 C178A

Though C178A is a point mutation, this is included in the list of structural mutants because it abolishes PrP’s disulfide bond. This mutant did not convert to PrPSc in ScN2a cells [Muramoto 1996] and did not cause any phenotype in mice [Muramoto 1997]. The susceptibility of the mice to prion infection was never tested.
MHM2 144#

This (along with the next entry) was the Prusiner lab’s attempt to create a mouse model of the Y145X mutation seen in humans (see above) and, simultaneously, to determine whether deleting both α-helices 2 and 3 would abrogate the neuronal storage disease phenotype. However, they never obtained cells nor mice with detectable expression of this mutant [Muramoto 1997].
MHM2 Δ23-88 144#

See MHM2 144# above. The Prusiner lab never obtained cells nor mice with detectable expression of this mutant [Muramoto 1997].
MHM2 Δ144-227

This mutant would have expressed the equivalent of the Y145X mutant but GPI-anchored instead of secreted. However, the Prusiner lab never obtained cells nor mice with detectable expression of this mutant [Muramoto 1997].
cyPrP

Ma Jiyan and Susan Lindquist deleted both the signal peptide (which directs PrP to the ER) and the GPI signal in order to create what they dubbed cyPrP – a version of MoPrP that is translated directly into the cytosol [Ma 2002a (ft)]. cyPrP was cytotoxic in neuroblastoma cells, but they did manage to create mice expressing the mutant, which became sick around 30 days of age and died around 70 days of age, due to atrophy of cerebellar granule cells. The original paper tied this observation into the Lindquist lab’s larger story about prion neurotoxicity being caused by blockade of the proteasome, and indeed, it was published back-to-back with another paper Ma 2002b (ft)] about how cytosolic accumulation of PrP due to proteasome inhibition leads to the rise of a PrPSc-like conformation. Ma has continued to study cyPrP since studying his own lab and now believes that the neurotoxicity of cyPrP requires that the hydrophobic core of PrP (amino acids ~112-134) be able to associate with the ER membrane [Wang 2006, Wang 2009].
MoPrP ΔGPI

A few different lines of mice have been created which express full-length PrP which lacks the GPI anchor and is thus secreted. The earliest reports had to be revised a few times [Chesebro 2005, Trifilo 2008, Chesebro 2010]. Secreted PrP appears to be preferentially degraded, making it hard to achieve an expression level comparable to wild-type PrP. At low expression levels, it is non-pathogenic, but when PrP ΔGPI is expressed in mice at > 1x levels, it spontaneously produces a lethal prion infection transmissible to wild-type mice [Stohr 2011]. The mice can also be infected with prions. Co-expression with wild-type PrP hastens the spontaneous disease, but co-expression with Δ23-88 does not.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.