Biolit 06: cytoskeletal dynamics
These are my notes for week 06 of Harvard’s BBS 230 “Analysis of the biological literature” course (October 7-9, 2014). This includes my own reading and analysis of the papers, as well as my notes from both discussion sections.
The papers for this week are Garner 2007 and Cao 2003.
Historical and scientific context
Decades ago it was thought that cytoskeletons were unique to eukaryotes. Eukaryotes have actin and tubulin, which differ in key ways. By 2004 it was known that prokaryotes had proteins with some functional resemblance to actin, but only limited sequence similarity. Dyche Mullins’s group had made major contributions to characterizing ParM, a prokaryotic actin homolog, and showing the ways in which it behaved differently than actin [Garner 2004]. In particular, the state of the ParM is determined by the balance of three forces that each differ from the equivalents for actin (differences in italics): ParM exhibits rapid (not just occasional) spontaneous nucleation, bipolar (not unipolar) elongation, and dynamic instability (as opposed to steady-state treadmilling).
Eukaryotic tubulin also exhibits dynamic instability, in contrast to actin. In eukaryotic mitosis, microtubules are thought to use “search and capture” to find chromosomes - they rapidly polymerize and depolymerize to find kinetochores to attach to.
Historically, the study of cytoskeletal proteins has relied on centrifugation to separate polymers (pellet) from monomers (supernatant), salt to induce polymerization and cold to induce depolymerization. Marine organisms (squid in particular) have been used as models because they have very long axons with very long actin filaments in them.
Garner 2007: Reconstitution of DNA segregation driven by assembly of a prokaryotic actin homolog.
Background
Plasmids are circular pieces of DNA, mobile genetic elements which bacteria replicate and distribute among their progeny. Note that for high copy number plasmids, the law of large numbers dictates that segregation between two daughter cells will be about 50/50. It is for low copy number plasmids (usually on the order of ~10 copies per cell) that it is very important for the DNA segregation mechanism to “get it right” - in particular, for two-copy plasmids, it is essential to transmit one copy to each daughter cell.
This paper uses the low copy number plasmid R1, which encodes ParC, ParR and ParM, as a model system to study the role of ParM, an actin homolog, in pushing plasmids apart after bacterial DNA replication.
The par operon in the R1 plasmid encodes three proteins:
- ParM is an actin-like filament protein
- ParR is a DNA-binding protein that must bind cooperatively in order for ParM to bind
- ParC’s DNA contains repeats that are the recognition sequence for ParR.
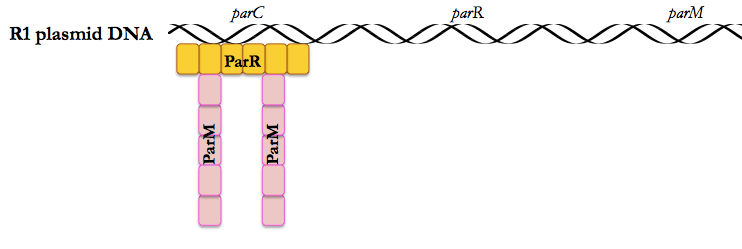
Mullins’ previous work had shown that (as stated above), ParM behaves differently from actin [Garner 2004]. Actin is stable, and the rate-limiting step in its formation is nucleation. In contrast, ParM undergoes frequent nucleation and polymerizes very quickly (it can extend the length of a bacterium in 10 seconds) but is also very unstable. Actin also requires a number of helper proteins (profilins for instance) to help it nucleate, etc., whereas you’d expect (and you’d be right) that because the R1 plasmid has to encode all the proteins needed for its own segregation, it must be a simpler autonomous system. This made it comparatively easier for Mullins to reconstitute in vitro. Even in a population of bacteria that all carry the R1 plasmid, only 40% will have ParM filaments at any given time, which means that these filaments are dynamic, assembled only when needed.
This paper relies on an in vitro reconstitution of the R1 system. This probably took a lot of debugging. The beads would need to be just the right size to be moved around by the ParM filaments, and all the surfaces involved would have to be the right materials so that the proteins don’t stick to them. The beads they used were 350 nm in diameter - larger and more massive than the R1 plasmid - and the length of ParM filaments they are able to form (ex. 10 μm in Fig 1G) are a few times longer than a bacterial cell, but the in vitro system also lacks many of the obstacles (other objects in the cytoplasm) that would probably hinder ParM filament elongation in vivo.
Figure by figure
Figure 1A-B show the formation of ParM asters around the beads. Figure 1C shows that a non-hydrolyzable ATP analog changes the dynamics, resulting in a smaller number of longer filaments. Figure 1D shows the “search and capture” mechanism of ParM filaments: if allowed to extend they will eventually find another bead. The remaining panels show the time series of how these filaments form (Fig 1F), the different shapes they can take on (Fig 1F vs. 1G), and what they look like up close (Fig 1E). Another subtle point about Figure 1G is that the triangle formed between 3 beads is equilateral - this hints at the bilateral elongation discussed more later.
Figure 2 shows what the “capture” event looks like (Fig 2A), that cleaving the filaments in the middle causes them to retract (not unlike a microtubule) (Fig 2B) and that the filament is able to find the longest dimension of an enclosure on its own (Fig 2C), implying that the mechanism for ensuring equal distibution of R1 copies between daughter cells is simply to push the R1 plasmids as far apart as possible so that they end up at opposite ends of the cell and will be split into different daughter cells upon mitosis.
In Figure 3, they use photobleaching to be able to monitor a spot on the filament to determine where monomers are being added.New monomers are added exclusively at either end of the filament - never in the middle. This means that the addition of monomers takes place at the interface between the filament and the plasmid. Fig 3C uses a technique called speckle microscopy, which was originally discovered by accident [Waterman-Storer 1998] - you add a substoichiometric amount of a fluorescently labeled protein, so that you can image individual fluorophores as points within a larger structure.
Figure 4A shows that the critical concentration of ParM required for sustained filament elongation is between 1.9 and 3.0 μM (they say 2.3 μM), with more limited elongation down to 0.6 μM. With a non-hydrolyzable ATP analog, ParM instead forms short, stable, non-dynamic filaments (Fig 4B). Addition of even a little bit of ATP (Fig 4C) is sufficient to make these filaments start to get longer, and the more ATP, the more rapidly they grow (Fig 4E). Together these findings suggest that ATP hydrolysis is required for elongation beyond a certain threshold, and that this ATP hydrolysis is promoted at the plasmid interface, which is what saves filaments that are bound to plasmids (or beads) from dynamic instability and catastrophe.
Cao 2003: The AAA-ATPase Cdc48/p97 regulates spindle disassembly at the end of mitosis.
Background
This paper uses a Xenopus egg extract system. For some facts and figures see Xenbase. Each Xenopus egg is >1 mm in diameter, visible to the naked eye, and can be synchronized or arrested in G2 mitosis which makes them really useful for studying development and cell cycle. Xenopus are tetraploid and have genomes about as large as humans (3e9 bp) so they are not especially preferred for genetics studies. Electric shock or addition of Ca2+ simulates fertilization and causes cell cycle to proceed.
An interesting nomenclature note is that many proteins such as p53, p97, etc. are named after their approximate size on a gel (e.g. p53 originally appeared to be ~53kDa in size). p97 is the frog ortholog of the Cdc48 protein from yeast - the authors refer to this single protein as Cdc48/p97.
By 2003, mitotic spindle assembly was reasonably well-studied, but next to nothing was known about spindle disassembly. Inactivation of Cdc2 (a kinase) at the end of mitosis leads to a cascade of dephosphorylation called the “mitotic exit network”, but whether (and if so how) this leads to spindle disassembly was unknown.
Figure by figure
In Figure 1 they add Xenopus sperm to egg extracts and compare the effect when p97 is or is not mutated to a dominant negative (p97QQ). The mutant does not affect spindle assembly (Fig 1A) but does prevent spindle disassembly (Fig 1B). The mechanism for the defect of p97QQ in Figure 1B is not that it prevents cell cycle exit as marked by decreased Cdc2 kinase activity (measured by cyclin B degradation and decreased phosphorylation of histone H1).
Immunodepleting the Ufd1-Npl4 complex (which interacts with p97) had similar effects as the p97QQ mutant: it allowed spindle assembly but prohibited disassembly, without affecting Cdc2 (Figure 2).
When centrosomes, rather than sperm, are used to induce spindle assembly in the extracts, inactivation of Ufd1-Npl4-p97 still blocks disassembly (Figure 3).
Figure 4 argues for a difference in microtubule kinetics upon perturbation of Ufd1-Npl4-p97, and Table 1 provides more quantitative data to back this up. The conclusion is that the rate of catastrophe is different between perturbed and control conditions.
Next they switch to a yeast system, and show that a mutant of the p97 homolog (cdc48-3) causes spindle collapse (Figure 5), a phenotype plausibly related to (though not identical to) the changes they saw in Xenopus extracts. Figure 6 and 7 present further evidence for Cdc48 regulating spindle disassembly, and presents a putative model where this occurs via Cdc5 degradation.
Discussion
Members of our class had a few critiques of this paper:
- All of the figures are cherry-picked single cells or single complexes - quantification is usually not performed (Table 1 is an exception) and statistical tests are never performed.
- They use a variety of methods to perturb the Ufd1-Npl4-p97 system (p97QQ, immunodepletion, antibodies against Ufd1 and other proteins), and the method they use is different for each figure.
- Better data visualization methods could help to make the take-home message clearer. For instance, Table 1 has a lot of rows and columns and a lot of leading zeroes, standard deviations are sometimes included and sometimes not, and one has to really squint at it for a while to see the core message (only fcat is different). Figure 5B presents as a series of histograms data which could be presented as a single plot of changing means with error bars over time.
Comparison
Garner 2007 uses a (comparatively) simple system, and one which was already sufficiently well understood to allow reduction to purified components in vitro. It presents a very organized series of experiments leading directly to conclusions. By the end of the paper, the fundamentals of how the ParM system works are very clear.
Cao 2003 is a more exploratory work. They are in a more complicated system/model (eukaryotic mitosis has to be more highly regulated and more complex than prokaryotic plasmid segregation because prevention of aneuploidy is so important) and the pathway they are studying (spindle disassembly) was largely unexplored prior to 2003. The interpretations of this paper are therefore by necessity more speculative. In addition, the experimental approach and data analysis have certain limitations as described above.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.