Biolit 10: metabolism
These are my notes for week 10 of Harvard’s BBS 230 “Analysis of the biological literature” course (November 18-20, 2014). This includes my own reading and analysis of the papers, as well as my notes from both discussion sections.
The papers for this week are [Yang 2002] and [Chen 2013].
General background
Here is cholesterol:
In animals, cholesterol inserts in membranes and reduces their fluidity as explained in Cell Biology 02: The Plasma Membrane. Instead of cholesteorl, fungi have ergosterol, plants have sitosterols. Cholesterol packaged into low-density lipoprotein (LDL) particles circulates in the blood. Increased LDL levels are strongly and causally associated with increased heart disease risk, as well as stroke. Increased high-density lipoprotein (HDL) levels in the blood are correlated with reduced heart disease risk, though there is not yet any evidence that this is causal, i.e. no evidence that genetic variants that increase HDL reduce heart disease risk.
A useful review of cholesterol metabolism is [Anderson 2003]; some of it is also discussed in Biochemistry 10: Lipid Metabolism. Prior to the [Yang 2002] paper below, the SREBP and SCAP proteins were known. In addition, it had been found that SCAP mutants were dominant negative: overexpression of these mutants resulted in cholesterol metabolism defects even if the wild-type protein was still present. This suggested that maybe the SCAP mutant depleted the available pool of some unknown co-factor. The major contribution of [Yang 2002] was to discover that this co-factor was INSIG-1.
Here is a diagram of some of the players in cholesterol metabolism:
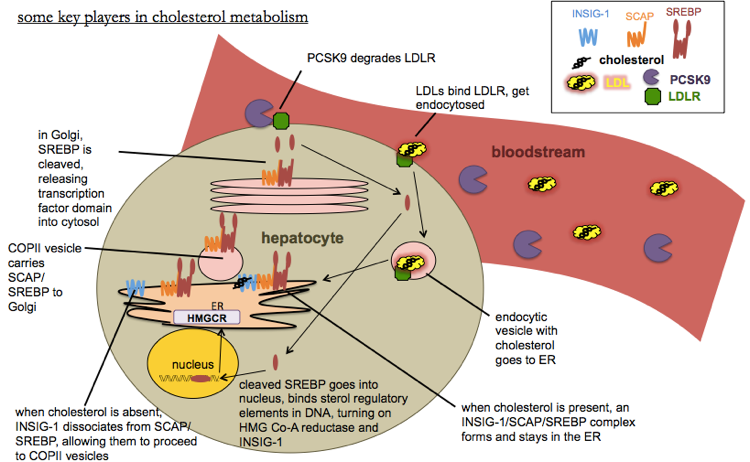
Cholesterol can be absorbed from the diet via NPC1L1 (NPC1L1), which is inhibited by ezetimbe, or it can be synthezied via the mevalonate pathway, the rate-limiting step of which is HMG Co-A reductase (HMGCR), which is inhibited by statins. To maintain cholesterol homeostasis, the cell regulates HMG Co-A reductase levels transcriptionally via sterol regulatory elements (SREs) in DNA. When cholesterol gets too low, INSIG-1 dissociates from the SCAP/SREBP complex, allowing the latter to move via COPII vesicles into the Golgi where SREBP is cleaved. The cleaved portion translocates to the nucleus where it acts as a transcription factor for SREs. This activates transcription of HMG Co-A reductase, which works to restore the minimum level of cholesterol. It also activates transcription of INSIG-1 itself, which acts as a negative feedback loop - making more INSIG-1 helps the SREBP pathway get shut off again once enough cholesterol is present, thus preventing an excess of cholesterol. The uptake of cholesterol into the cell from the bloodstream is performed by LDLR, which is negatively regulated by PCSK9.
Over 30 million Americans take statins.
Yang 2002: Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER
This paper reports the discovery of a novel protein dubbed INSIG-1, which in humans is encoded by the INSIG1 gene.
Figure by figure
They began by expressing in HEK293S cells FLAG-tagged versions of SCAP containing (TM1-6) or lacking (TM1-5) a putative sterol-sensing domain thought to interact with the hypothesized co-factor (see above). They then performed co-immunoprecipitation and mass spectrometry to try to identify the proteins that uniquely bound to the version containing the binding domain. They found 7 proteins, of which they chose one for further followup because it was found in mouse livers overepressing SREBP1.
Figure 1 characterizes the newly discovered protein in CHO cells. (Aside: they switch between a lot of different types of cells in this paper and it is unclear why). They characterize its sequence (Fig 1A), a hydropathy plot (Fig 1B), and its expression across different tissues - it is mainly expressed in liver (Fig 1C). It also provides a highly unconvincing microscopy image to show that the protein localizes to the ER whether or not sterols are present (Fig 1D).
Figure 2 characterizes the relationship between INSIG-1, SCAP and SREBP - particularly, under which conditions SREBP gets cleaved and translocates to the nucleus. In the absence of SCAP (first two lanes of Fig 2A), SREBP-1a never gets cleaved, regardless of whether INSIG-1 is present. When SCAP is available, SREBP-1a is readily cleaved in the absence of INSIG-1 and its cleavage is dose-dependently inhibited by INSIG-1. SREBP-2 behaves similarly (Fig 2B). They considered vesicular stomatitis virus G protein (VSVG) as a control Golgi protein and confirmed that INSIG-1 does not affect VSVG localization to Golgi and, by implication, is not a general regulator of ER→Golgi traffic.
They hypothesized that INSIG-1 negatively regulates SREBP cleavage by causing SCAP to be retained in the ER. The proteases that cleave SREBP are presumed to be located in the Golgi. Therefore, if you could retain those proteases in the ER, you could get cleavage back, even in the presence of INSIG-1. Therefore Figure 3 uses Brefeldin A, a small molecule inhibitor of GBF1 which results in retention of Golgi proteins in the ER. And sure enough, Brefeldin A rescues the inhibition of SREBP cleavage by INSIG-1. This means that INSIG-1’s mechanism of action is upstream of the need for transport to the Golgi.
Figure 4 performs gel shift assays to determine what physically interacts with what. It appears that INSIG-1 binds SCAP, but only in the presence of sterols. Figure 5 confirms this by showing a dose dependency in the amount of unbound INSIG-1 and the amount of SREBP cleavage. The gels and the densitometric quantification of them (Fig 5C) are messy, yet the message comes through:
SREBP-2 cleavage is blocked by sterols only when there is excess unbound INSIG-1 available so that it can bind all of the SCAP/SREBP-2 complex.
Figure 6 confirms that a sterol-insensitive mutant of SCAP is indeed deficient at binding INSIG-1.
Figure 7 confirms that the presence of INSIG-1 alters the dose dependency between sterol levels and SREBP-2 cleavage.
Figure 8 further shows (lane 4) that if you leave out SCAP, there is no INSIG-1/SREBP complex. This disproves a direct INSIG-1/SREBP interaction.
Epilogue
This paper was seminal, establishing the role of INSIG-1 in sterol regulation. It leaves some questions open - for instance they can’t rule out that there may be other players as well. How can you rule out other players? It would be almost impossible to reconstitute the whole system in vitro (since the proteins are membrane-bound), so instead you could try to reconstitute it in a very different organism (maybe yeast) that you expect does not have endogenous copies any of the other putative members of the complex. This was a useful approach for other pathways - for instance, yeast do not have RNAi, so David Bartel used yeast to reconstitute the RNAi pathway.
Chen 2013: SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion
Historical and scientific context
You can get cholesterol one of two ways: absorption from the diet, or de novo synthesis. Both of these pathways have been drugged: ezetimibe inhibits NPC1L1, reducing absorption of dietary cholesterol, while statins inhibit HMG Co-A reductase, reducing synthesis of cholesterol. In spite of this, millions of people still have high cholesterol, and heart disease is the largest cause of death in the developed world. Therefore, there is a continuing medical need to reduce circulating LDL.
As chronicled in this primer, human genetics have established PCSK9 as a pretty much ideal target for reducing LDL. Yet PCSK9 has so far proven impossible to drug with a small molecule. It’s not clear why this is, since there exist other serine protease inhibitors - perhaps it’s because PCSK9’s catalytic activity is not actually required for LDLR degradation [McNutt 2007]. But in any case, both of the anti-PCSK9 drugs nearing FDA approval (evolocumab and alirocumab) are monoclonal antibodies. mAbs are expensive to produce, and have to be admininstered intravenously, which makes these drugs somewhat less likely to be blockbusters like statins. This is perhaps what motivates an interest in other targets further upstream which regulate PCSK9 but might prove more amenable to small molecule inhibition.
This paper discovers that Sec24a regulates PCSK9 secretion, and that Sec24a knockout mice have reduced circulating PCSK9 and reduced LDL. It’s not clear if they had any idea from the outset that their studies of Sec24a would lead them to PCSK9, but in any case, it ended up being the main message of the paper.
In yeast, Sec24 knockout is lethal. Humans (and mice) have four paralogous copies of it, though, Sec24a-d, which allows more robustness and allows each paralog to regulate different cargoes. SEC24A in ExAC has several loss-of-function variants, though there are no homozygotes.
Figure by figure
This figure describes how they used the “gene trap” method (see the genetics 201 lecture on zebrafish) to knock out the SEC24A homolog in mice, and provides various lines of evidence that the gene was indeed knocked out and that the mice are healthy and viable.
This table simply provides evidence that the breeding outcomes were Mendelian, consistent with the knockout mice being having no defects in viability or fertility.
This table lists the values of a bunch of different blood phenotypes in Sec24a knockout mice. The authors’ goal is to show that there is no difference in any of these phenotypes between knockout and wild-type. Some of the differences are statistically significant, but only one survives multiple correction testing, and all of the differences are so small in magnitude as to be negligible.
Fig 2 describes how they made additional conditional knockout alleles of Sec24a in order to be sure that the viability they saw was not a result of some residual amount of protein.
Fig 3 shows that the knockout mice have hypocholesterolemia. APOA1 is depleted in the blood of knockout mice (Fig 3B-C); this protein is normally found in HDL but is not a specific marker - it is also found in some other lipoprotein particles. Fig 3D shows that both HDL and LDL are reduced in the knockouts. The effect is not sex-dependent (Fig 3E-F) nor specific to one knockout allele (Fig 3G).
In class we debated how meaningful these ~50% reductions are. What is the relevant dynamic range of blood lipids? I didn’t find any figures for mice. I did find Mayo Clinic’s blood lipid guidance for humans, according to which the difference between “ideal” and “very high” LDL is about a 2x difference (100 mg/dL vs. 190 mg/dL respectively). So if mice are anything like humans, a 50% reduction in LDL in the knockout mice is indeed a pretty “clinically significant” reduction.
As a rescue experiment, they used FLP to “flip” out the gene trap, thus restoring a functional gene product. This successfully rescued the knockout phenotype (Fig 4A-B).
Sec24a is expressed in many tissues, and the authors wanted to know which tissues were important for the cholesterol phenotype. They hypothesized the liver was important. To be very specific about this, one might express Cre under a liver-specific promoter to knock out Sec24a only in the liver. The authors did not do that, though - instead they intravenously injected an adenoviral vector containing Cre under an EIIA promoter. This would be expected to knock out Sec24a in a subset of cells in a variety of tissues - neither the intravenous injection nor the promoter (EIIA) are liver-specific. The authors claim they have evidence that the Sec24a expression in the liver is what affects cholesterol, but their data do not actually support this.
The foregoing results indicated that Sec24a regulates cholesterol metabolism, but it wasn’t clear how. One possibility was that it affected the SREBP pathway we discussed in reference to the previous paper. If that were true, you’d expect a whole range of SREBP targets to be downregulated. Therefore this figure characterizes transcriptional differences in wild-type vs. knockout livers using RNA-seq plus qPCR on a selected subset. Though there are some significant differentially expressed transcripts, they don’t coalesce into any clear pattern in GO terminology, nor do they significantly suggest a disruption of SREBP signaling. There are also Western blots to show that knockout does not affect SREBP cleavage.
Consider that even if Sec24a knockout doesn’t directly affect the SREBP pathway, you could still imagine it would affect SREBP just by affecting cholesterol. But in this case, apparently that effect is not detectable. I guess if Sec24a knockout reduces PCSK9 secretion, that would increase LDLR’s uptake of LDL into the cell, so you’d expect the SREBP pathway to be inhibited (i.e. SREBP targets turned off) more than in wild-type mice, even while circulating LDL is reduced. But the reduction in circulating LDL may in turn lead to slightly less cholesterol in the cell, so perhaps the steady-state level of SREBP pathway activation is the same. Or the difference is just not detectable in their blots.
They next wanted to know if the cholesterol deficiency was due to less production, or higher clearance, of cholesterol. Apolipoprotein E is involved in clearance of cholesterol from plasma, and Apoe knockout mice have high cholesterol levels in their blood (the same is true of an APOE knockout human reported earlier this year [Mak 2014]). The researchers therefore crossed their Sec24a knockouts with Apoe knockouts and homozygosed both alleles to create double null mice. The double null mice had about the same cholesterol levels as Apoe null mice did. This means Apoe is epistatic to Sec24a, implying that the mechanism by which Sec24a works must relate to receptor-mediated clearance of cholesterol. In further support of this, Ldlr + Sec24a double knockout mice also had the same phenotype as Ldlr knockout mice. Thus, Sec24a knockout must reduce Ldlr- and Apoe-mediated clearance of cholesterol from blood. They then used ELISA to measure circulating levels of PCSK9 and found they were reduced 55% in the Sec24a knockout mice. This makes sense because LDLR clears cholesterol and PCSK9 acts by degrading LDLR.
This figure provides evidence that Sec24a knockout results in a reduction in transport of PCSK9 from the ER onward to the Golgi.
Endo H is a glycosylase that cleaves off immature glycan chains found in the ER, but is unable to cleave glycan chains that have matured after spending some formative time in the Golgi. In Sec24a knockouts, the PCSK9 shifts its electrophoretic mobility upon Endo H treatment, indicating that its glycans never matured - from this they conclude that PCSK9 is being retained in the ER in the absence of Sec24a.
Brefeldin A inhibits transport into the Golgi. It inhibits secretion of PCSK9 (Fig 7C), demonstrating that PCSK9 goes via the Golgi (how is this a surprise??). Fig 7D shows that a battery of different treatments that inhibit COPII transport inhibit PCSK9 secretion, and also that PCSK9 and Sec22b (a known COPII substrate) behave the same as one another. Together these facts argue that PCSK9 transport is COPII-dependent.
Aside: by this point in the paper, the authors have shifted their focus entirely to PCSK9 and are not pursuing any other possible substrates of Sec24a transport. What would you do if you wanted to know what all other proteins are trafficked by Sec24a? One possibility would to perform proteomics on COPII vesicles isolated from wild-type versus Sec24a knockout cells. No one in our class knew exactly how to isolate COPII vesicles, but it was speculated that perhaps you could lyse cells in a hypotonic solution and then use an antibody to COPII to pull down the vesicles.
Because Sec24a, b, c and d are all paralogous, they wanted to know which one(s) account for the most transporting of PCSK9. They do this by knocking down each of the different genes in rat hepatoma cells and then performing an in vitro COPII vesicle budding assay. They find that Sec24a does the most transporting, and Sec24b does some; Sec24c and d do not have appreciable roles.
Following up on the in vitro experiments from Fig 8, they crossed the Sec24a knockout mice or wild-type mice to heterozygosity for a Sec24b knockout allele. This further reduced cholesterol in the mouse bloodstream on the Sec24a knockout background but did not have an appreciable effect on a wild-type background.
Epilogue
This paper establishes viability of Sec24a knockout and clarifies some of how PCSK9 is transported. In terms of medical relevance, it’s hard to believe that Sec24a is not responsible for transport of any other cargoes. Presumably it transports a lot of substrates, and so one might expect that inhibition of it would have a lot of different undesirable side effects. So Sec24a is unlikely to become a new drug target for PCSK9 reduction.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.