Protein folding 07: Collagen
These are my notes from week 7 of MIT course 7.88j: Protein Folding and Human Disease, held by Dr. Jonathan King on March 19, 2015.
Assignment 6
The readings for this week are chapter 14 of Branden & Tooze, and [Kuivaniemi 1991, Laganowsky 2012, Schmid 1995].
Kuivaniemi 1991
Many forms of osteogenesis imperfecta (OI) are caused by missense substitutions of glycine for something else. Amazingly, many of these are lethal in a heterozygous state. Because collagen folding is processive from C terminus to N terminus, a single missense substitution is sufficient to disrupt not just a local folding environment but the folding of the entire chain. Also surprisingly, missense variants that abolish prolines are less associated with disease.
Schmid 1995
In addition to prolyl hydroxylase (discussed below), it turns out that prolyl isomerase is also important for collagen folding. Collagen translation is a few times slower than the translation of the average eukaryotic protein, which gives enough time for prolines to cis-trans isomerize before translation is complete. Yet collagen folding requires 100% trans proline. Thus, it has been proposed that prolyl isomerase is required to maintain the trans-ness of prolines in collagen, though this hasn’t been definitively proven yet. Eventually this enzyme turned out to be important for many other proteins too. Some pharmas consider prolyl isomerases to be potential targets for anti-inflammatory drugs, though this approach could lead to adverse effects on collagen throughout the body.
Laganowsky 2012
For years, amyloid fibrils were thought to be the neurotoxic species in neurodegenerative disease. In recent years, attention has shifted towards lower molecular weight oligomers. This paper examines how such oligomers could be cytotoxic. They produce a crystal of cylindrin, which forms a hexameric cylindrical barrel made of antiparallel beta strands [Figure 2]. They propose that the mature amyloid is parallel in-register, and that the transition from the hexameric precursor into mature amyloid requires breaking the hydrogen bonds between strands. However, the energetic barrier to this is extremely high, and they don’t offer any explanation as to what interacting partner or biological process drives this proposed refolding event.
Lecture: collagen
Collagen is found in all metazoan animals, and in mammals, it is 20% of total protein. It is not found in plants, which have cellulose instead. It is has many roles:
- Major structural component of tendons and cartilage
- Confers mechanical strength of skin and cornea
- Constrains arteries and veins against the force of blood pressure
- Forms the scaffold that shapes bones and teeth, filled in with mineral deposits
- Critical in wound healing
Collagen, isolated from whatever tissue in whatever animal, forms macroscopic fibers banded at intervals of 670Å. Unlike muscle they are not compressible nor extensible. Instead they are more analogous to girders in construction. They are thought to have evolved to withstand the force of gravity. They are secreted proteins which assemble fibers in the extracellular space. Although cells do bind to the collagen fibers, it is more appropriate to think of collagen fibers as operating at a tissue level rather than a cellular level.
Collagen is the molecular basis of leather, most glues up until modern times, and gelatin. These are three materials with very different physical properties. In fact, collagens have diverse properties in vivo too, conferred in part by different amino acid sequences. Most mammals each have about 20 paralogous collagen-like genes. The most abundant is type I collagen which is involved in tendon, ligament, bone and tooth formation. Also noteworthy is type IV collagen, which gives mechanical strength to veins and arteries.
Type I collagens are ~1054 residues long and have a very distinctive amino acid sequence, with 1/3 of all residues being glycine, and high levels (~10% in humans) of proline and hydroxyproline. Hydroxyproline is made by a post-translational modification thanks to the enzyme prolyl hydroxylase. A typical sequence motif is GXPGPYGP-Hyp where Hyp stands for hydroxyproline which appears not to have a one-letter acronym. Collagens also contain hydroxylated lysines which participate in covalent cross-links. Tryptophan, tyrosine and cysteine are extremely rare in collagen, with the result that collagen is virtually invisible in terms of 280nm absorbance. Thus, even if you think you’ve purified DNA and have removed all protein because there is negligible 280nm absorbance, you can’t rule out considerable amounts of collagen, which after all was probably 20% of the original protein. Collagen is also highly protease-resistant and cannot be broken down by trypsin or chymotrypsin. It’s very difficult to get collagen to break down into monomers in order to study it in a molecular biology context. When we eat meat, the combination of cooking to denature the collagen, plus low pH in gastric acids, allow us to digest it. Bacteria that grow on rotting meat produce an enzyme called collagenase which can break it down.
The study of collagen was traditionally undertaken for industrial rather than scientific ends. And because it forms fibers and not crystals, it took many decades before structural biologists recognized collagen as being a considerable part of our knowledge of protein-folding. In recent years, collagen has re-entered the biomedical realm to some extent due to interest in inflammation.
Fibroblasts are among the cells that migrate to close a wound. They secrete collagen even when grown in tissue culture, despite the lack of any bone or other tissue for the collagen to bind to.
Alex Rich and Francis Crick solved the structure of collagen by X-ray fiber diffraction [Rich & Crick 1955, Rich & Crick 1961], in close competition with Ramachandran’s group, which arrived at the same structure. It is a left-handed helix with 120° rotation per residue and a 2.86Å rise per residue (compare to 100° and 1.5Å in alpha helices). Groups of three helices are wrapped around each other in a right-handed fashion. The carbonyl oxygens and amide nitrogens are orthogonal to the fiber axis (compare to alpha helices, where they are parallel to the fiber axis). The reason why every third residue has to be a glycine is that the 120° turn is such that every one of these glycines points to the same side, to the interface with another helix, and the absence of a side chain allows closer packing. Each triple helix is about 1000 amino acids, or 2800Å long. The three chains each have tripeptide repeat motifs, but their residue numberings are staggered, such that the repeat starts at residue 1 in one chain, residue 2 in the next, and residue 3 in the third chain. The frequent prolines have two contradictory effects. On one hand, they remove a degree of rotational freedom (φ), making the chain stiffer in the Cα-N bond. However, the prolines also remove an amide proton, with the result that the adjacent N-C bond lacks double bond character, and is thus more rotationally free, leading to cis-trans isomerism. Hydrogen bondong between helices is mediated by amide protons of glycine on one chain to a carbonyl oxygen of a proline on the next chain. There is only one hydrogen bond per three residues - much less than in alpha helices or beta sheets. Thus you cannot attribute the extraordinary stability of collagen to an extensive hydrogen bonding network.
In human tendons and ligaments, the bundle of three helices is composed of two copies of collagen 1A and one copy of collagen 1B, however, any one collagen peptide is capable of forming a homotrimeric triple helix. The diversity in collagen genes is not necessary for triple helix assembly but is needed for higher-order assembly of macrofibrils.
Helen Berman, a major force behind PDB, has managed to crystallize collagen [PDB# 1CGD, Bella 1995], and found that the crystals have the same structure that Rich & Crick found by X-ray fiber diffraction:
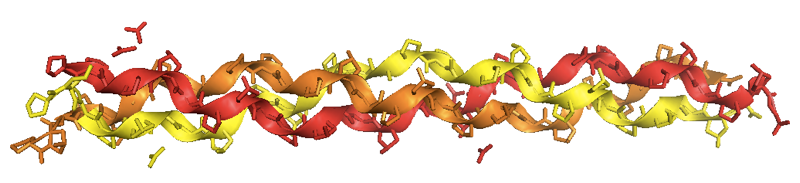
In the collagen fibrils of all known species, individual collagen fibers are packed into macrofibrils with a stagger of about 1/4.5, meaning that if each fibril has length L, their starts occur at intervals D = L/4.5. Every 5 fibers are packed into a fibril, and those fibrils are then packed next to each other in a supertwisted fashion. The “stagger” of the molecules gives rise to the banding patterns observed in macrofibrils. In the macrofibrils, the molecules are covalently cross-linked to one another. The degree of cross-linking increases over time, and in fact part of the reason that old people get wrinkles is that over time the collagen in skin becomes more and more cross-linked such that it is difficult for the body to break it down.
The molecular biologist who wishes to study collagen wants to obtain newly synthesized collagen monomers before they become cross-linked into a macrofibril. To obtain monomers, you have to use tissues that are growing rapidly, thus where the cells are rapidly producing new collagen. Therefore collagen is often prepared from embryonic chick tendon, or skull calvaria which is patches of bone that spread as the head grows and then knit together once the animal has grown enough, or baby rat tail. You can mince the tissue and extract it in high salt which removes the already-soluble blood and lymph proteins. These procedures leave cells and soluble proteins intact. You spin the cell debris down and retain the pellet, which contains the collagen that is still in the secretory pathway inside the cell. You then resuspend the pellet in acetic acid at pH 3, which will denature most other proteins but leave collagen intact. We now spin again, and the denatured proteins will pellet out, while the collagen will be soluble. You reserve the supernatant and dialyze it to restore it to low salt and neutral pH. Collagen fiber precursors will spontaneously form in this solution. You then stain these fiber precursors with uranyl acetate. The “uranyl” is a uranium ion; large atoms like uranium absorb X-rays such that dense absorbent pockets on your sample represent gaps in the protein structure where the uranyl acetate has filled in a hole.
Hodge & Schmitt followed this procedure and found that the protofibrils formed sheets 3000Å wide but of variable length. These turned out to be triple-helical molecules that had aligned with each other in register, without staggering [Hodge & Schmitt 1960]. These triple-helical molecules have a particular circular dichroism spectrum, distinct from that of alpha helices, which you can use to monitor their folding. With this preparation of collagen molecules, Hodge & Schmitt then used heat to study equilibrium folding and unfolding of collagen. In equilibrium unfolding, the molecules begin in 300 kDa in-register trimers with helical CD absorption, and at around 40° they melt into 100 kDa monomers and thereby lose their helicity. Thus, folding is cooperative. In equilibrium folding, however, they found that once unfolded by heat the molecules could never fold again, and instead formed gelatin.
Polyglycine and polyproline, like collagen, will each form left-handed triple-helical fibers with intermolecular hydrogen bonds. Synthetic peptides with sequences like GAPGPP also do this, and these have been used to study the relationship between length and stability. The longer the monomers, the higher the thermal stability. Also, the higher percentage proline, the higher the stability. This relationship persists across collagens from diverse species, and it turns out that the proline effect is due to conformational entropy. Because of the conformational stiffness of prolines, an unfolded collagen monomer is much longer (more extended) than any other polypeptide of comparable length. Because the monomer is so extended, the interaction that leads to a trimer is more entropically favorable. Another way to think about this is that every tripeptide that lacks a proline is not stiff and so is able to open up and nucleate an unfolding event across the chain.
If you examine collagens across species, there is an approximately linear correlation between environmental temperature (body temperature for warm-blooded animals, ambient temperature for cold-blooded animals), and the melting temperature of their collagen.
Hydroxyproline is found almost exclusively in triple helix domains in animal proteins. Because the proline has a ring and conformationally constrains the chain with or without a hydroxyl group, the only difference appears to be that hydroxyproline can bind a water molecule. In fact, it appears that hydroxyproline organizes a cage of water molecules that surround the triple helix and also bind to one another [Bella 1994, Bella 1995]. Collagen had been used for years to form a thin matrix on film that held the silver nitrate for analog photographs, and engineers in the photography industry had noticed that collagen held water, and that it lost its desirable properties if dehydrated. When triple helices are packed together with other triple helices, it is actually the water shells, not the proteins themselves, that contact one another.
The catalytic activity of prolyl hydroxylase requires iron, and anemia actually leads to defects in hydroxlation of prolines on collagen, and subsequent collagen defects. Cultured fibroblasts secrete triple helices - the collagen monomers actually associate with one another before leaving the cell. If you grow the cells with iron chelators, you can deprive them of hydroxyproline, and thereby measure the thermal properties of collagen with different degrees of hydroxylation of proline. If you plot the hydroxyproline:proline ratio, you find that at 50 you get a Tm of 38°C and at 10 you get a Tm of 22°C.
Certain aspects of genetic collagen disorders can be understood directly from the structure of collagen. This is unique - even for hemoglobin, we can only understand experimentally, and not intuitively, why certain missense variants disrupt the fold.
Human diseases caused by collagen defects include:
- Osteogenesis imperfecta. This is the most common birth defect, which presents as congenital big bone deformations readily recognized at birth by obstetricians.
- Marfan syndrome
- Ehlers-Danlos syndrome
- Scurvy, believe it or not
- Many others [Kuivaniemi 1991]
Marfan syndrome and Ehler-Danlos syndrome are actually caused by genetic defects not in collagen itself, but in its interacting partners.
Scurvy was first observed during long-haul nautical trips by European explorers. It presents with chronic bleeding, starting with the gums. Wounds don’t heal, and you die of internal bleeding. This is because prolyl hydroxylase actually requires several things: iron, O2, alpha-ketoglutarate, and ascorbic acid. When you lack ascorbic acid, your collagen gets depleted for hydroxyproline, which reduces its thermal stability. Thus, when fibroblasts try to heal a wound, the collagen just melts. Analogous to scurvy, there are certain grasses that contain iron chelators, and livestock that graze on these plants get iron deficiency resulting in collagen defects.
Gelatin occurs when a large collection of collagen molecules form local triple helix interactions but out-of-register such that three whole molecules do not align together, and instead each molecule binds to many others. Given that gelatin forms upon cooling of a heated collagen mixture, how do animals prevent newly synthesized collagen molecules from forming gelatin in vivo?
An elegant experiment answered this question using collagen from rat calvaria [Bellamy & Bornstein 1971]. The primary cells were incubated with tritiated (3H) proline as a pulse-chase. Cells were synthesizing collagen at about 200 residues per minute, or ~6 minutes per chain. They then did autoradiograms at defined time intervals to characterize the “hot” prolines fresh off the ribosomes. The earliest-detected hot species was of very high molecular weight (not a single chain). Its molecular weight was reduced somewhat by protease treatment and reduction by β-mercaptoethanol. If you waited longer before collecting the hot collagen, you would also get lower molecular weight species. When they examined the earliest species, they found that it contained a dense mat surrounded by a fuzzy “fringe” which was accessible to proteases and reducing agents. It turned out that the earliest species was made of trimers of procollagen, which retained N- and C-terminal prodomains. These prodomains did not have collagen-like repeats, and instead were rich in cysteine and were disulfide bonded to one another. These prodomains were dubbed “registration sequences”. The maturation from procollagen to collagen was slower in type IV than type I collagen, so they focused their efforts on type IV. They found that the registration peptides had to recognize each other and force the collagen repeats to get into register and form a triple helix. Surprisingly, the folding proceeds from C terminus to N terminus rather than vice versa. Prolyl hydroxylase can only modify exposed, not triple helically folded, prolines, and it appears to proceed from C to N terminus ahead of the folding frontier. The procollagen trimers get secreted as-is, and in the extracellular environment, they encounter procollagen peptidase which removes the prodomains. Procollagen peptidase is often expressed and secreted by different cell types than the ones that produce collagen - Bellamy and Bornstein’s primary cultures didn’t express it, which is why they were able to detect the prodomains. Some cattle have a skin disease called dermatospataxis, caused by genetic defects in procollagen peptidase.
Tanning of leather involves making the cross-links between collagen triple helices resistant to degradation by bacterial and fungal collagenases.
Addendum from lecture 8
These notes were added from the April 2, 2015 lecture.
One more important experiment about collagen folding [Harrington & Rao 1970] deserves mention.
Collagen in vitro refolding is studied by heating the protein to melt it, then performing serial dilutions at the high temperature, then lowering the temperature to 30°C to allow re-folding. This was originally done using “optical rotatory dispersion” (ORD) instruments, which apparently no longer exist, having been replaced by circular dichroism (CD), which follows the same principles. After lowering the temperature, you measure the re-attainment of helical structure as measured by ORD (y axis), over the course of tens of minutes (x axis). They then varied the concentration of collagen. Because 3 chains have to associate, you might hypothesize that the refolding rate would be proportional to the 3rd power of the collagen chain concentration. But in fact what they found is that the refolding curve was almost unchanged between .002, .004, and .008 g/mL. Only when you raise the concentration to .016 g/mL do you finally see an increase in the refolding rate. The authors did not have an explanation for this phenomenon.
Later on, an explanation was uncovered. Collagen is >1,000 residues long and composed of triplet repeats. The helicity measurement obtained by ORD is only measuring local helical structure and can tell you nothing about the superstructure. It turns out that at low concentrations, helicity was dominanted by intramolecular helix formation - the protein curls around on itself twice and forms a triple helix with itself. Only when you cross a particular concentration threshold do the molecules find each other at a significant enough rate to be able to form intermolecular triple helices. Even then, they are forming gelatin with local alignment but lacking proper global alignment (because they are mature collagen peptides lacking the registration domains).
They also looked at the initial rate of refolding vs. temperature, over a range of temperatures below the melting point, from about 10°C to 30°C. In other proteins, it was known that refolding proceeds faster at higher temperatures, provided you are still below the melting point. But surprisingly, collagen refolded most quickly at the low end, around 10°C, and refolded much more slowly at 30°C. This turns out to be explained by a nucleation and growth model. Collagen folding is processive once three chains have come together, but the initial “nucleus” - the initial point of association of the three chains in the proper helical conformation - is highly thermolabile. So at temperatures closer to 30°C, the nucleus is less likely to survive long enough to nucleate a full refolding event.
An interesting contrast is that in collagen we are confident that the “nucleus” has the same conformation as the rest of the chain eventually will after folding proceeds. For amyloid nucleation models, we do not know whether the initial nucleus of misfolding has the same conformation as the final amyloid fiber does.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.