Thalidomide's renaissance
In the second episode of Breaking Bad, Walter White teaches his chemistry class about chirality:
For instance, thalidomide. The right-handed isomer of the drug thalidomide is a perfectly fine, good medicine to give to a pregnant woman to prevent morning sickness, but make the mistake of giving that same pregnant woman the left-handed isomer of the drug thalidomide, and her child will be born with horrible birth defects. Which is precisely what happened in the 1950s. So: chiral, chirality, mirrored images - right? Active, inactive, good, bad.
Walter White’s summary isn’t quite correct — (R)- and (S)-thalidomide readily interconvert in vivo [Teo 2004], such that (R)-thalidomide would not actually be a “perfectly fine, good medicine” for pregnant women — but thalidomide is certainly the classic example of the starkly different bioactive properties that two entantiomers can possess. Aside from its role in introductory chemistry classes, the drug is also invoked as a cautionary tale supporting careful regulation of pharmaceuticals: the FDA never approved it for morning sickness, ensuring that its European legacy of birth defects never extended to the U.S. But one way or the other, the tragedy of the 1950s is the only context in which most of us have ever heard of thalidomide.
Yet over the past few months, I find myself hearing the word thalidomide more and more, and this time people are talking about it for a different reason: its unique mechanism of action.
Thalidomide has already enjoyed a second life for almost two decades. In 1998, it received FDA approval for the treatment of a complication of leprosy, and in 2006 it was approved for multiple myeloma as well. Now marketed as Thalomid® in the U.S., thalidomide has been earning $245 million per year in revenue for Celgene.
And all of that happened without us having basically any idea what its mechanism of action was, until a few years ago.
In the first breakthrough, thalidomide was conjugated to beads, the beads were incubated with cell lysates, and the proteins that stuck to the beads were subjected to mass spectrometry. Two proteins were specifically pulled down by thalidomide and were revealed to be cereblon (CRBN) and damaged DNA binding protein 1 (DDB1) [Ito 2010]. Cereblon was shown to be the direct target, and DDB1 indirect, as thalidomide pulled down recombinant cereblon on its own but only pulled down DDB1 if cereblon was also present. CRBN was at that time already known as a recessive intellectual disability gene [Higgins 2004], though the function of its protein product was unclear. DDB1, however, was known to be a component of E3 ubiquitin ligase complexes [Angers 2006], and the authors were able to show that cereblon, DDB1, and another protein called Cul4a formed a complex with autoubiquitination activity - an activity which was inhibited by thalidomide [Ito 2010]. Thus, thalidomide’s target was cereblon, and the drug also affected the E3 ubiquitin ligase activities of its larger complex.
By screening a battery of deletion mutants and point mutants of cereblon, they identified a rough binding site for thalidomide and two point mutations — Y384A and W386A — that together virtually abolished thalidomide binding. Thalidomide was teratogenic in zebrafish and chick embryos, as it is in humans, but expression of Y384A/W386A double mutant cereblon rescued this effect in both species, thus demonstrating that thalidomide’s binding to cereblon was the basis of its teratogenicity [Ito 2010]. The choice to use fish and chicks for these experiments was critical because thalidomide has long been known to lack teratogenic activity in mice [Fratta 1965], a subject I’ll return to later in this post.
Within a couple of years, another group showed that the two thalidomide analogs lenalidomide and pomalidomide, FDA-approved for various blood cancers, also bind to cereblon [Lopez-Girona 2012]. Just as with thalidomide, these drugs inhibited the autoubiquitination of the cereblon-DDB1 complex, and the Y384A/W386A double mutant was resistant.
But did thalidomide and its analogs achieve their effects by abolishing some native function of cereblon, or by conferring upon it a new or heightened function? Ito et al were of two minds on this subject, at one point asserting that “thalidomide exerts teratogenic effects by binding to CRBN and inhibiting its function,” but also noting that “Whereas CRBN is ubiquitously expressed in humans, thalidomide exerts tissue-specific effects. Evidently, CRBN is necessary, but not sufficient, for thalidomide teratogenicity” [Ito 2010]. Ito had shown that thalidomide could inhibit an autoubiquitination activity, and that thalidomide caused some phenotypes in zebrafish that were similar to those caused by knockdown of Cul4a, another member of the complex [Ito 2010]. Yet in the later study, when cancer cell lines were chronically exposed to lenalidomide or pomalidomide, they developed drug resistance through decreased cereblon expression [Lopez-Girona 2012], which would suggest the drugs conferred a gain of function upon cereblon. Human genetics didn’t provide a smoking gun answer to the gain-vs-loss of function question either. The fact that mutations in CRBN seem to cause mild intellectual disability rather than birth defects [Higgins 2004] would have argued against simple inactivation of cereblon by the drug being responsible for teratogenicity, except that the causal mutation in the family Higgins studied was a stop codon very near the end of the protein (R419X in a protein of just 442 residues), where a total loss of function is by no means guaranteed. And as of this writing, in the 11 years since Higgins’ paper, no other pathogenic mutations in CRBN have been reported in ClinVar.
Nonetheless, after a couple of years, three groups using different methods arrived almost simultaneously at the same answer for these drugs’ mechanism of action [Gandhi 2014, Kronke 2014, Lu 2014]. When lenalidomide binds to cereblon, it increases the E3 ubiquitin ligase complex’s affinity for two transcription factors, Ikaros (encoded by IKZF1) and Aiolos (encoded by IKZF3), leading to the ubiquitination and proteasomal degradation of those proteins. This was discovered through a variety of approaches. Kronke and Gandhi had each begun by performed quantitative mass spec proteomics, while Lu had created an arrayed library of 15,000 proteins fused to luciferase. All three found that these two proteins were among those most strongly reduced in abundance when cells were treated with lenalidomide. Lu and Kronke each then showed that Ikaros and Aiolos form a classic ubiquitination “ladder” on a Western blot when cells are treated with lenalidomide, while Gandhi used cycloheximide pulse-chase to show that lenalidomide reduced the half-life of the two proteins. All of the groups showed that siRNA knockdown of IKZF1 or IKZF3 could mimic some of the effects of lenalidomide in cell lines. By creating a series of deletion mutants of Aiolos (IKZF3), Kronke et al identified a 59-residue susbtring of amino acids that was sufficient to cause a protein to be ubiquitinated by cereblon in the presence of lenalidomide [Kronke 2014]. Within this sequence, dubbed a “degron”, the sensitivity of IKZF1 and IKZF3 appears to be attributable to a single amino acid difference (Q instead of H) versus IKZF2 and IKZF4, which are not affected by lenalidomide.
So why does ubiquitination and degradation of Ikaros and Aiolos result in any therapeutic value against myeloma? It was known that thalidomide induces expression of the cytokine interleukin-2, thus potentially hastening an immune response to cancer [Haslett 1998], and that Ikaros and Aiolos are transcription factors that repress, among other things, interleukin-2 [Thompson 2007, Quintana 2012]. Thus, Lu speculated, thalidomide and its analogs, by causing Ikaros and Aiolos to be degraded, might be freeing interleukin-2 from transcriptional repression, triggering a protective cytokine response. Kronke went a step further, at least partially confirming this hypothesis by showing that lenalidomide induces IL-2 mRNA, and that siRNA against IKZF1 or IKZF3 does the same, and that the combined effects of lenalidomide and the siRNA are less than additive, suggesting that the same mechanism is at play.
That same year, two groups solved the structure of thalidomide and/or lenalidomide bound to cereblon and DDB1 [Fischer 2014, Chamberlain 2014]. One of these structures [PDB# 4TZ4] is shown below:
fetch 4tz4
bg_color white
hide everything
show surface, chain A or chain C
color 0xFF9912, chain C
color 0x0000CD, chain A
show sticks, organic
color 0x000000, organic
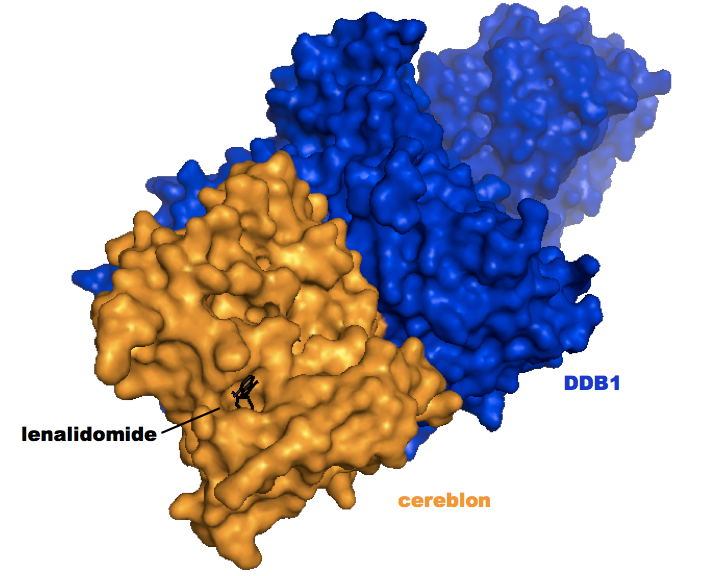
Zooming in, lenalidomide almost appears as a tiny black key in a lock:
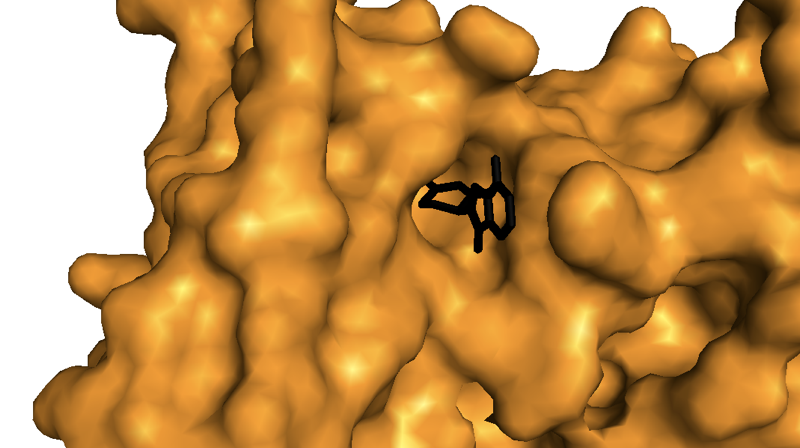
Indeed, the cleft that the drug binds on the protein’s surface is so deep that when I play with the structure in PyMOL, it’s actually hard to find an angle where you can get a clear view into it. Is the presence of such a binding pocket on this E3 ubiquitin ligase complex just a convenient coincidence, or did cereblon evolve to bind some endogenous small molecule? We don’t yet know. Kronke points out one precedent for an endogenous small molecule regulator of ubiquitination — auxin, a growth hormone in plants, is known to bind to a ubiquitin ligase and change its affinity for various substrates too [Tan 2007]. A recent commentary [King & Finley 2014] points out that there is also a precedent in mammals: FBXL3, a ubiquitin ligase that targets cryptochromes, thus controlling the circadian clock, is regulated by flavin adenine dinucleotide [Xing 2013]. But if cereblon has an endogenous small molecule ligand, no one has found it yet.
Another curiosity about the structure is that one group reported a structure for mouse cereblon bound to thalidomide [Chamberlain 2014, PDB # 4ZTC]. But wait, I thought thalidomide didn’t cause birth defects in mice — otherwise it would surely never have been prescribed to pregnant women? Yet thalidomide does bind its target in mice? Yes, apparently. Indeed, not one of the amino acids that physically forms the thalidomide binding site differs between mouse and human.
This inconsistency led another group to construct a battery of mouse/human cereblon chimeras to narrow down the region of the protein responsible for the species difference, and then to introduce each mouse-human amino acid difference in that region, one by one, to see which made the difference [Kronke & Fink 2015]. The answer was human cereblon residue V387, which is I in mouse cereblon. When a V387I mutation (which at least a few humans actually have, by the way) was introduced into human cereblon, it became unresponsive to lenalidomide; when the converse mutation, I391V in mouse numbering, was put into mouse cereblon, it became responsive to lenalidomide. The only structural difference that the V387I mutation introduces appears to be that the extra methyl group on the isoleuceine protrudes as a tiny bump on the surface of the protein just outside of the lenalidomide binding site [Kronke & Fink 2015] — shown here on structure 4ZTC:
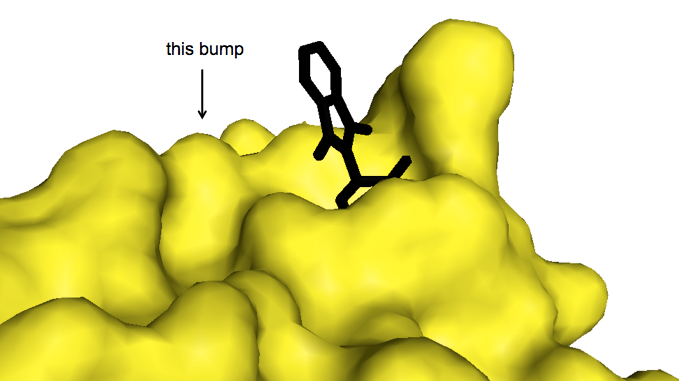
Though this bump apparently does not preclude lenalidomide (nor, presumably, thalidomide or pomalidomide) from binding, it must have some subtle yet profound effect on the efficiency of the protein-protein interaction interface between cereblon and its substrates. When I said in the last paragraph that the V387I mutation made human cereblon unresponsive to lenalidomide, that is true both for degradation of Ikaros and Aiolos, and also for degradation of another protein called casein kinase 1α, which is the main subject of that paper [Kronke & Fink 2015]. That group had just identified yet a third protein, known as CK1α for short (and encoded by CSNK1A1) which lenalidomide causes cereblon to target for degradation. The degradation of CK1α is apparently responsible for yet another clinically useful pharmacological activity of lenalidomide, which is its efficacy in myelodysplastic syndrome with a hemizygous deletion of chromosome 5q — yet another type of hematological cancer. And CK1α, the authors show, does not have the same “degron” amino acid sequence that Ikaros and Aiolos share, but instead must be recognized by cereblon through some other sequence.
This expansion of the set of lenalidomide’s indirect targets to include CK1α offers an enticing hint that cereblon’s activity is not limited to just a couple of transcription factors relevant to myeloma — it can be pharmacologically induced to target other proteins too, who knows how many. Indeed, it’s already known that thalidomide reduces TNFα expression [Sampaio 1991], an activity which we must now consider might be mediated, however indirectly, by binding to cereblon as well. And for all the inroads into understanding the anticancer effects of thalidomide and its analogs, we still don’t know which, if any, of these molecular outcomes actually account for why thalidomide causes birth defects†, or why it alleviates morning sickness. There may be yet another substrate, or many substrates, of cereblon’s ubiquitin ligase complex, that account for these other effects.
†Update 2018-08-13: a new paper Donovan 2018 now reports that SALL4 is the cereblon target that mediates the birth defects caused by thalidomide.
But the ability of thalidomide to cause cereblon to degrade several different proteins is only exciting if we get to choose which proteins it degrades. A drug that hits several targets is a drug that’s likely to be — indeed, was, and is — quite toxic, at least in some settings. But it turns out that CC-122, an experimental analog of thalidomide now in clinical trials, causes degradation of Ikaros and Aiolos but not CK1α [Kronke & Fink 2015].
So if drugs can be designed to lend cereblon specificity for just one or a few proteins, then the door is wide open to imagining what could be done with this mechanism. And indeed, a lot more than imagining has been going on. A few months ago, in almost perfect sync, two different groups published their own bifunctional molecules that hijack cereblon by conjugating thalidomide to a small molecule ligand of BRD4, a bromodomain protein that reads activating acetylation marks on chromatin (see these notes) and is important for the growth of some cancers [Winter 2015, Lu 2015].
One molecule, dubbed dBET1, successfully reduced BRD4 protein levels by up to 80 or 90% (see Fig 1J, with a sub-micromolar EC50 [Winter 2015]. It worked in vivo, too, reducing tumor size in mice. And the molecule was fairly specific: according to their proteomic analyses the molecule resulted in reduced levels of three other bromodomain proteins (which of course is an issue of the bromodomain ligand’s specificity, not cereblon’s specificity), and just two other proteins, MYC and PIM1, at least one of which is in fact a desired downstream target of BRD4 inhibition [Zuber 2011]. The other group’s molecule appeared to be at least as efficacious, with an EC50 of ~1 nM and a pronounced effect on cancer cell line growth [Lu 2015].
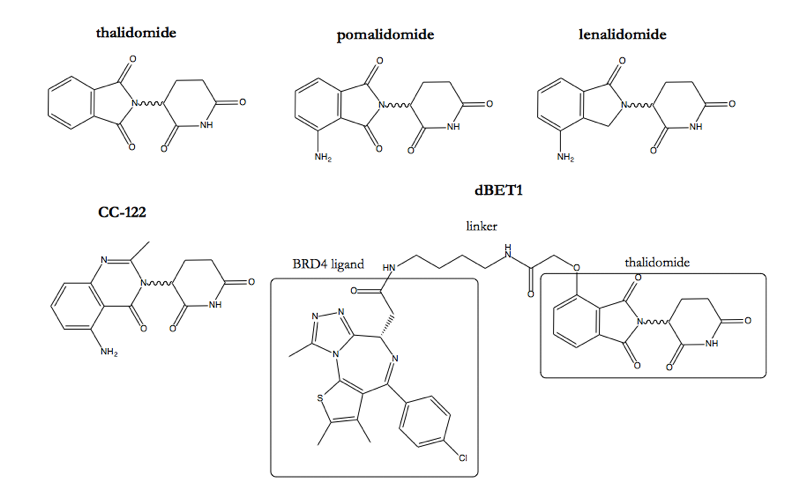
And notice that the BRD4-binding bifunctional molecules are a whole conceptual leap away from the original series of thalidomide anlaogs. Whereas the original molecules just slightly alter a protein-protein interaction surface, with apparent effects on its affinity for multiple substrates, these bifunctional molecules go out and just wholesale grab onto a different protein, possibly one which in nature would never have interacted with cereblon to begin with. So in principle, it seems possible to achieve a great deal of specificity with this approach. If you already have a protein you know how to bind with a small molecule, but you don’t know how to degrade it, just stick it onto thalidomide:
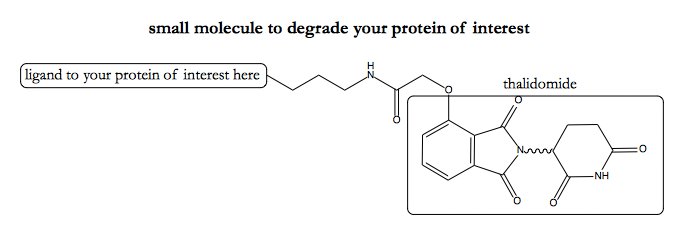
This, at last, must be why I’ve been hearing so much talk about thalidomide lately. The hope here is that maybe we’ve hit upon a modular way to target any protein for degradation, analogous to, say, targeting any mRNA for degradation using an siRNA of the right sequence.
As usual, the reality probably won’t be quite as rosy as the hype. In an enthusiastic-but-also-critical blog post, Derek Lowe points out three potential problems — finding a good ligand to your protein of interest in the first place, linking it to thalidomide without losing either one’s activity, and then doing the medicinal chemistry to allow the resulting molecule to get into the cytosol.
And indeed, isn’t another requirement of this strategy that your protein of interest be cytosolic in the first place? Consider trying to apply this strategy to PrP, which is a secretory pathway protein. It begins its life being co-translationally translocated into the ER, transits through the Golgi, and emerges, GPI-anchored, to divide its time between the cell surface and the endosomes. Never in this journey is it in the same subcellular compartment as cereblon, so even if you had a good PrP ligand (which I’m not sure we do), where is the opportunity to trigger degradation? Purely for the sake of illustration, let’s suppose that the cationic porphyrin FeTMPyP binds PrPC, as reported [Nicoll 2010], and that it can be conjugated to thalidomide via a linker without losing activity. PrP binds one end of the bifunctional molecule, and cereblon binds the other, but never do the two bind the same copy of the molecule:

Perhaps I’m wrong about this — if so, I’d like to see what exactly this diagram looks like for a scenario where the molecule does bring PrP and cereblon into proximity. (Here’s the original if anyone wants to edit).
Now, granted, some groups have reported seeing some PrP in the cytosol some of the time under some circumstances [Zanusso 1999, Jin 2000, Yedidia 2001, Ma & Lindquist 2001] — see this post for a review — and UPS reporter mice [Dantuma 2000], whose brains express GFP under conditions of proteasomal stress, glow in the terminal stages of prion disease [Kristiansen 2007]. So it’s not as though PrP has never been sighted in the same compartment as cereblon. But to the extent that there exists such a thing as cytosolic PrP, is it a large enough fraction of total PrP to be worth targeting? And isn’t it in the cytosol precisely because it has already been targeted for degradation via ERAD? And if so, is there any marginal benefit to ubiquitinating it even more?
For these reasons I am skeptical that the thalidomide conjugation approach could be relevant to PrP, which is my protein of interest. Of course, the thalidomide concept is just the latest technology in a series of bifunctional molecule concepts. Some of these concepts are very similar and suffer from the same limitations — for instance, over a decade ago, there was an effort to use bifunctional small molecules to deliver your-protein-of-interest to Skp1-Cullin-F box, another E3 ubiquitin ligase complex [Sakamoto 2001], and a couple of years ago, a patent was filed on an approach to do the same for VHL, the ubiquitin ligase that’s mutated in von Hippel Lindau [CA2861066A1]. Then there are the bifunctional molecules to dimerize a protein or bring your-protein-of-interest into proximity with something else of interest. For instance, a molecule consisting of two copies of FK506 fused together, dubbed “FK1012”, can be used to trigger FKBP12, and other proteins bound to it, to dimerize [Spencer 1993]. Or, in the neurodegeneration space, there was SLF, another FKBP12 ligand, fused to Congo red, which commandeered FKBP12’s steric bulk to block the elongation of Aβ fibrils [Gestwicki 2004].
Perhaps the approach most specifically relevant to secretory pathways proteins is the idea of adding a bulky hydrophobic group to a ligand to attract the ER protein quality control chaperone BiP. BiP recognizes exposed hydrophobic patches, which usually indicate protein misfolding, and it can then help with folding or direct proteins for degradation [Kleizen & Braakman 2004]. The first proofs of principle for this strategy were done in pretty artificial systems: the target was a protein of interest genetically fused to a HaloTag protein, and the ligand was a covalent ligand of HaloTag, fused to some collection of aromatic rings or cyclohexanes or an adamantane as the hydrophobic moiety [Neklesa 2011, Tae 2012]. It was later shown that similar results could be achieved with a non-covalent ligand [Long 2012; and see commentary in Neklesa & Crews 2012], though still in an artificial system, targeting a bacterial protein for degradation in mammalian cells. Just recently, success was reported in designing a molecule, dubbed SARD279, consisting of an androgen receptor (AR) ligand, fused via a linker to an adamantyl group, to cause AR degradation, with 50% degradation achieved at 1 μM [Gustafson 2015].
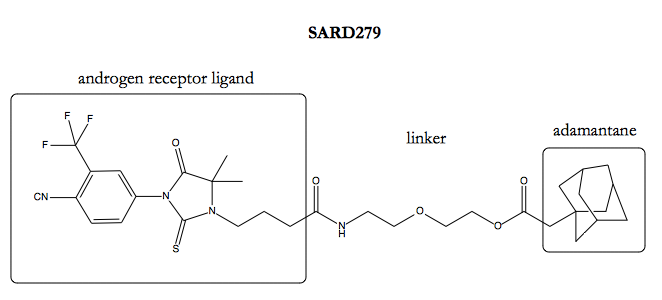
Whether any of these approaches will yield a drug remains to be seen. It’s an area that deserves both excitement and, as Derek Lowe points out, some dose of cautious skepticism. So perhaps what’s most exciting about the emerging thalidomide story, then, is simply the proof that using a small molecule to trigger a protein’s degradation isn’t an impossible concept for a drug. In fact, unwittingly, we’ve already been doing it for years.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.