Small molecules to reduce one protein's expression
introduction
This post will explore strategies to use small molecules to reduce the expression of one protein, at any point up to and including its translation. By way of introduction, it’s important to discuss why such strategies might be of interest.
My protein of interest is the prion protein, PrP. Reduction of PrP expression has an extremely strong proof of principle as a therapeutic strategy. Knockout mice are resistant to prion disease [Bueler 1993], and incubation time is inversely correlated with PrP expression level all the way from heterozygous knockouts to 10x overexpressers [Bueler 1994, Fischer 1996]. Cre-mediated conditional knockout of PrP around the time of first symptoms can reverse prion disease [Mallucci 2003], and conditional downregulation to ~20% of wild-type levels using a Tet-off system can dramatically delay prion disease [Safar 2005]. So: if you can shut off production of the the protein, you can stop the disease. But how to achieve that?
In a recent post about thalidomide I discussed strategies for targeting one protein for degradation after it has been translated and has folded. So why not just focus on those strategies? There are a few reasons.
First, the strategies discussed in that post all require having a good small molecule ligand for the protein of interest to begin with. For PrP, it’s not clear whether we have such a ligand available. The same is true for many proteins implicated in human disease.
Second, the strategy of bringing your protein of interest into proximity with an E3 ubiquitin ligase, is probably only relevant to cytosolic proteins. PrP is an extracellular protein, GPI-anchored to the plasma membrane and localized to the cell surface via the secretory pathway. (The alternative strategy for secretory pathway proteins may be adamantyl conjugation to attract BiP. There are now some promising results for using this strategy to induce androgen receptor degradation [Gustafson 2015], so I think it is worth pursuing — if a ligand can be found.)
A third issue, which someone pointed out to me recently, is that even if you have a small molecule ligand for your protein, it probably binds to the protein in its natively folded state. For a disease, such as prion disease, where the problem is one of protein aggregation, do we know that individual PrP molecules reach a fully folded PrPC state before being recruited to PrPSc? Or is it possible that some nascent PrP molecules never even fold correctly in the first place before becoming PrPSc? The fact that GdnHCl is useful in getting PrP to convert to a PrPSc-like conformation in various in vitro paradigms [Kocisko 1994, Atarashi 2011] leads us to consider the possibility that any PrP molecules that never reach their full native fold may be preferentially converted to PrPSc. I speculate that this might be particularly true in genetic prion disease if the pathogenic mutation causes PrP to take longer to fold — though mutant PrPs can reach a fold nearly indistinguishable from the native fold of wild-type PrP [Lee 2010], I don’t know if the kinetics of their folding have been extensively studied.
And finally, antibodies to some epitopes of PrP’s globular domain can be toxic [Sonati 2013]. It is not yet known whether small molecules that bind PrP might have similar effects, and I certainly still think that small molecule binders of PrPC are worth pursuing, but the possibility of epitope-specific toxicity is at least one reason for considering alternative approaches in tandem.
To be clear, loads of drugs have effects on gene expression, and there exist loads of data about this — that’s what LINCS is all about. In some instances, therapeutic applications of compounds have been suggested specifically on the basis of their effects on transcription — examples, such as celastrol, are discussed in this lecture and this one. But this post is about what to do if, like me, you have exactly one protein whose abundance you want to alter, while affecting everything else as little as possible. To figure out if there are ways of specifically altering one protein’s expression level up to and including translation, Sonia and I both looked to the literature and sat down to brainstorm with our colleagues at the Broad Institute to see if they knew of any good examples. Here’s everything we found, broken down by the process being targeted.
transcription
![]()
Above: some small molecules that affect transcription through epigenetic changes or transcription factor destruction.
We didn’t find any clear examples of small molecules specifically targeting one gene or a few genes’ transcription. Often, strategies for affecting transcriptional regulation are epigenetic — say, targeting chromatin state with HDAC inhibitors, or BET bromodomain inhibitors [Filippakopoulos 2010, Matzuk 2012], or, rather than inhibiting an epigenetic protein, targeting it for destruction [Winter 2015]. Then there is the strategy of targeting transcription factors for destruction (lenalidomide’s mechanism). All of these approaches will affect expression levels of tons of different genes. As far as I can tell, such molecules are being pursued therapeutically in instances where dysregulation of the transcription factor or epigenetic regulator itself is a cause of disease, rather than just to get at one gene that happens to be regulated by them.
One slightly odd example of a more targeted attempt is the effort to disrupt HIV Tat protein’s interaction with the HIV TAR RNA stem-loop. Sonia came across a screen of tripeptides for the ability to bind the HIV-1 TAR RNA stem-loop [Hwang 1999]. Evidently the HIV Tat protein must bind this stem-loop structure, and Tat is important for the processivitity (ability to continue transcribing once it has started) of the RNA-dependent RNA polymerase that makes HIV mRNA from HIV genomic RNA. After screening a combinatorial library of 24,389 tripeptides against TAR, they found a few with sub-micromolar KD. One of these was shown to inhibit expression of a reporter gene with an EC50 of about 50 nM. All of those results sound awesome, but there must be some catch or else this would be a drug by now. Though they counterscreened for cytotoxicity, the compound was only incubated with the cells for 4 hours, so maybe it later turned out that it was incredibly non-specific and just killed transcription globally or something. If anyone knows the epilogue on these tripeptides I would be curious to hear it.
splicing
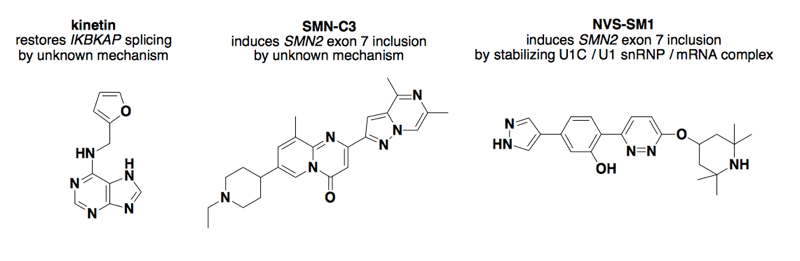
Above: some small molecules that affect splicing.
Therapeutic alteration of splicing is a sufficiently hot area that there was a whole review on it a few years ago [Spitali & Aartsma-Rus 2012]. Almost all of that review is about antisense oligonucleotides, which are not small molecules and are thus beyond the scope of this blog post. The only small molecule example mentioned therein is kinetin.
Kinetin is a natural product marketed as an active ingredient of anti-aging creams — you can even buy it on Amazon Prime. In a screen of just 1,040 known bioactives, kinetin was found to restore proper splicing of IKBKAP, the gene disrupted in familial dysautonomia (FD) [Slaugenhaupt 2004]. Sue Slaugenhaupt later filed a patent on the use of kinetin and related analogues to treat FD [US20150111902]. The compound has been shown to improve splicing in a mouse model of FD [Shetty 2011] and has gone to clinical trials [Axelrod 2011, NCT02274051] but the mechanism of action is apparently still unknown.
Recently there have been two breakthroughs in finding small molecules to alter splicing of SMN2 to treat spinal muscular atrophy (as also mentioned in this post). In the first instance [Naryshkin 2014], the mechanism was not identified. The latter compound was shown to stabilize a transient complex of the U1 snRNP, the U1C protein, and the nascent mRNA during splicing.
How specific are these compounds? Both studies performed RNA-seq oncultured cells treated with their compounds to see how many genes were differentially expressed in the presence of their compounds. The first study found only 12 genes whose expression was either increased by ≥2x or decreased to ≤0.5x in the presence of the compound [Naryshkin 2014]. The other found 175 genes differentially expressed at this same threshold, plus 39 differential splicing events [Palacino 2015]. They explored the sequence motif required for a splice site to be affected by the compound, and evidently the affected splice sites were enriched for an unusual nGA sequence motif present in SMN2 exon 7 but only 2.6% of all human exons. So that fact that SMN2 happens to have a relatively rare splice site sequence may be part of what allowed this small molecule to be relatively specific, affecting its splicing without disturbing too many other genes. The compound (Novartis LMI-070) is now in a Phase 1 clinical trial for SMA [NCT02268552], so we’ll soon know how well-tolerated it proves to be in humans.
Kinetin and LMI-070 are both intended to restore expression, of a mutated or pseudogene transcript respectively. We didn’t find any examples of drugs targeting splicing specifically to disrupt one protein’s expression. But looking at Figure 3A of [Palacino 2015], you can see there are some transcripts whose expression in log2 space drops below the -5 mark, implying a >97% decrease in expression upon treatment with LMI-070. In this case, where the goal is to change SMN2 splicing, those are considered off-target effects. But if one of those transcripts happened to encode the protein that causes your gain-of-function disease, then you’d have quite a potent drug candidate on your hands. Thus I infer from this example that in principle, it may be possible to knock down a gene of interest by targeting its splicing.
Just a note, these few examples discussed above are not a complete list. A series of other natural products and synthetic derivatives are known to inhibit splicing factor 3b (encoded by SF3B1), with possible relevance to treatment of some cancers [Kotake 2007, Kaida 2007, Albert 2009], and there are several other splice-inhibiting compounds reviewed in [Bonnal 2012].
mRNA stability
Obviously, there are non-small-molecule approaches for targeting mRNA — siRNA and antisense oligonucleotides can both induce mRNA destruction. In addition, the reason that some of the splice-modulating drugs discussed above cause a decrease in expression of some transcripts might be that they cause inclusion of cryptic non-conserved exons that contain stop codons, thus triggering nonsense-mediated decay, so these might indirectly affect mRNA stability. However, we did not find any good examples of small molecules that bind directly to an mRNA and cause it to be degraded. In fact, one review [Thomas & Hergenrother 2008] specifically states that there are no examples in this area.
translation initiation
![]()
Above: some small molecules that target translation.
The first concept we ran across is that of riboswitches: structures in RNA that evolved to bind small molecules. Apparently these are prevalent in bacteria as mechanisms for small molecule metabolites to regulate their own production pathways, and some of them are now believed to be the targets of both synthetic and natural product antibiotics such as roseoflavin [reviewed in Blount & Breaker 2006, Serganov & Patel 2007]. Though this is very cool, it doesn’t yet seem to amount to a more general proof of principle that you can target a specific RNA with a small molecule. After all, the whole strategy is predicated on your target of interest already having a binding pocket, which won’t usually be the case. And I am not aware of any riboswitches encoded in the human genome.
Another strategy for targeting translation initiation is to go after the initiation factors themselves rather than the mRNA. One study specifically developed a FRET screen to try to prevent formation of a productive translation initiation complex involving eIF4GI and eIF4E [Cencic 2011] (see background in molecular biology 27). The paper doesn’t go into a ton of detail about why eIF4E would be a good drug target. They just note in the introduction that disruption of the eIF4F complex, of which eIF4E is part, has “modest effects” on global translation rates and disproportionate effects on a few mRNAs, particularly those with more secondary structure. Mice tolerated 15 mg/kg/day of their lead compound, 4E1RCat, for 5 days, so it at least wasn’t acutely toxic. They tested it as a cancer treatment and reported that it was ineffective alone but had some synergy with doxorubicin. I would be interested to see some ribosome profling or proteomics data on cells treated with this compound to see how specific its effects are.
Novartis once did a screen for inhibitors of IRES-mediated translation [Didiot 2013]. IRES are internal ribosome entry sites, structures in mRNA which allow translation to begin while bypassing most of the cell’s usual translation initiation machinery (see molecular biology 27 for background). Because many viruses, and apparently the oncogene c-Myc, use IRES, while most eukaryotic transcripts do not, this might be a good drug target. They found two hits, cymarin and somalin, which had about 20-fold selectivity for inhibiting IRES over regular cap-dependent translation (for instance, somalin inhibited cap-dependent translation with an EC50 of 2 μM but inhibited IRES-dependent translation with an EC50 of 100 nM). That’s probably nowhere near the specificity you’d want for a drug, but maybe it’s a start. The compounds killed c-Myc-dependent cancer cell lines, suggesting possible therapeutic utility. I would be interested to know if Novartis is still exploring analogues of these compounds or, generally, pursuing this target at all.
The “iron response element”, discovered decades ago [Hentze 1987], is a well-defined stem-loop structure in the 5’UTR of some mRNAs which iron response protein 1 (IRP1) binds in order to regulate translation. There have been a few efforts to find ways of targeting iron response elements’ activity, whether by directly binding the mRNA or through indirect mechanisms. One screen found a compound that appeared to increase IRP1 interactions with a iron response element, but the direct mechanism of action was not clarified [Zimmer 2008]. Another group fused the 5’UTR of APP mRNA to a luciferase open reading frame, and screened for compounds that would reduce translation of luciferase. They reported several hits, at least one of which, JTR-009, was claimed to directly bind the iron response element in the mRNA [Bandyopadhyay 2006, Bandyopadhyay 2013].
As an aside and a cautionary tale about screening, a colleague also pointed us to an interesting story where a group had tried to screen for compounds that increase translation [Shin 2014]. Their hits, hymenialdisine and isohymenialdisine, turned out to be inhibitors of translational repression by PKR — evidently, the addition of mRNA to in vitro extracts for screening caused this translational repression response, and the hits were simply counteracting that response.
discussion
If you want to target one gene’s expression with a small molecule, the better proofs of principle seem to be for going after splicing or maybe translation initiation. We didn’t find very good of examples targeting transcription or mRNA stability. Maybe we just missed something in the literature — leave a comment to let us know.
If there’s a generalizable lesson from the examples reviewed above, it might be that if you want to target one gene’s expression, you need there to be something special about that gene. A rare splice site sequence, or a rare translation initiation mechanism, maybe a rare RNA secondary structure, something that your gene of interest has that relatively few genes in the human genome have. To the extend that any of the examples reviewed above have demonstrated specificity, it’s through targeting that which is rare. And even then, none of the examples above seem to have specificity anywhere near that of small molecules directly targeting proteins of interest.
With this in mind, an upcoming post will discuss PrP’s life from transcription initiation to translation, and whether there is anything rare or unique about it that could be targeted to reduce its expression.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.