Read with caution! This post was written during early stages of trying to understand a complex scientific problem, and we didn't get everything right. The original author no longer endorses the content of this post. It is being left online for historical reasons, but read at your own risk. |

This post has been deprecated. It was written when the author was just beginning a scientific career and may not necessarily represent an informed scientific understanding. It has been left online for historic reasons. Read at your own risk.
Walker, Wedemeyer and Scheraga, in A role for intermolecular disulfide bonds in prion diseases? (2001, in PNAS) hypothesize that the intramolecular disulfide bond between C179 and C214 in PrPC is replaced in PrPSc with an intermolecular disulfide bond. If true, this would provide a mechanism for PrPSc propagation– two PrP molecules form an intermolecular disulfide bond, leaving their remaining cysteines as thiol (-SH) groups which are free to “attack” other PrP molecules, breaking up their intramolecular disulfide bonds and adding them to the oligomer. This not only sounds plausible, it also explains why higher PrPC levels make an organism easier to infect (see blog entry on Walker Jackson’s FFI mice), and can also explain some otherwise unexplained aspects of prion diseases:
- The high temperatures required to destroy prion infectivity. The authors note that this is easier to account for with a covalent chemical bond than if the infectivity were just based on hydrogen bonding supporting a particular protein conformation.
- The connection between metal ions and prions. The authors note that the octapeptide repeats are metal ion-binding and that extra repeats can cause prion disease (but there are all sorts of other connections between prions and copper and manganese too)
- In the authors’ words: “The cellular form PrPC also has a remarkable conformational stability in the vicinity of the disulfide bond (21) and an unusually fast folding time (26), which may derive from the need to stabilize the critical intracellular disulfide bond against thiolate attack.”
Here’s one other hypothesis I’ll also throw out randomly, don’t know if it’s valid or not: this could also go at least
some length toward explaining why there are different strains of prion diseases, a phenomenon for which I’ve not yet heard any other satisfactory explanations. If you’ve got cysteines at codon 179 and codon 214, then there are three possible intermolecular disulfide bonds you could form: 179-179, 214-214 or 179-214. Then you could imagine one kind of aggregate consisting of PrP molecules bound 179-179 and 214-214, or an aggregate consisting of molecules all bound 179-214 one to the next. Each of these forms would have the same protein-based infectivity that is unique to PrP, because each would have a thiol group free to attack PrPC and add it to the aggregate; yet each could produce a distinct protein conformation leading to different parts of the molecule being exposed and therefore different types of toxicity. This could help to explain the three phenotypes of heritable prion diseases in humans– CJD, FFI and GSS– (and
some researchers say vCJD and kuru constitute fourth and fifth distinct non-hereditary phenotypes).
Short of designing an experiment and heading to the lab to test Walker et al’s hypothesis, it’s tempting to see what other data out there we can correlate with what they’ve noted. Walker et al make the claim that “many of the mutations that provoke inherited prion diseases are clustered near the (intramolecular) PrP
C disulfide bond” and cite
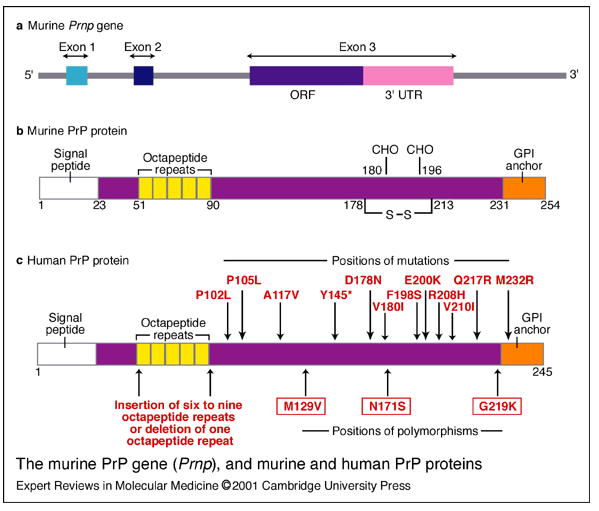
Wadsworth for this. The implication is that hereditary prion diseases consist of mutations that weaken the conformational support for the intramolecular disulfide bond, thus making it more likely to come undone, or more vulnerable to attack by other thiols. Here is Wadsworth’s diagram of the location of PrP polymorphisms and mutations:
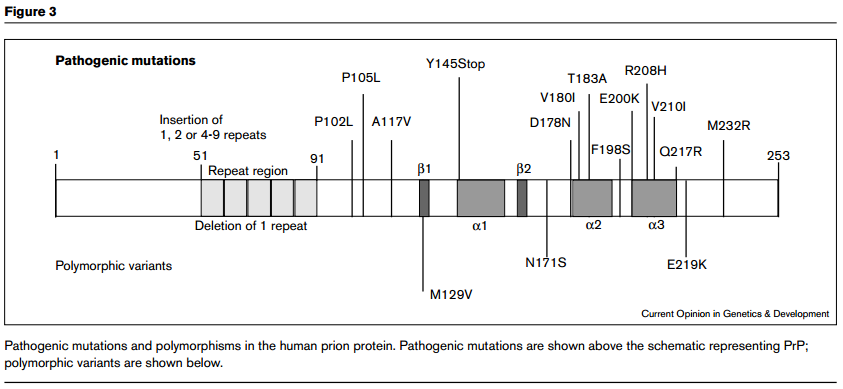
I wish that these diagrams provided more obvious support for Walker, Wedemeyer and Scheraga’s claim. Yes, D178N and
V180I are both disease-causing mutations and are just left and right of the C179 involved in the disulfide bond. But then there’s F198S, which causes GSS, and it’s right in the loop region between α-helices 2 and 3 (i.e. NOT right next to a cysteine) and then there are the octapeptide repeat deletions or additions, which are way far away from the cysteines. And the would-be smoking guns of mutations at 213 and 215 to match 178 and 180 are not to be seen. Compare the above diagrams to Riesner’s diagram of PrPC structure:
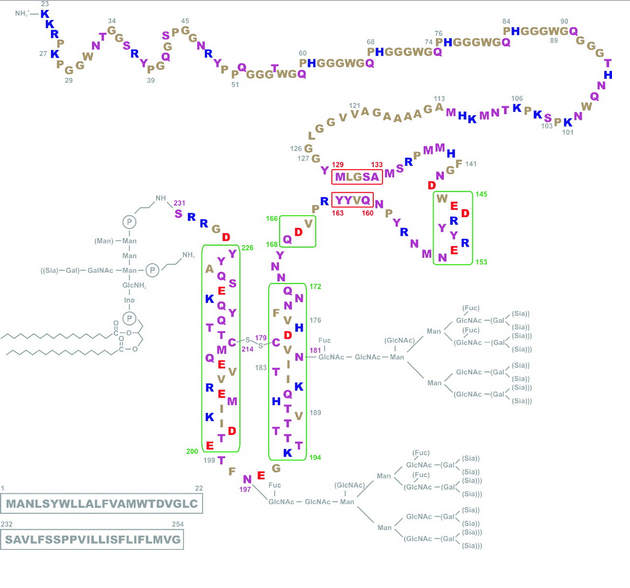
Suddenly 105, 117, 198 and others all look rather far from the disulfide bond action. But this isn’t to say the hypothesis is wrong (far be it from me to critique the work of real scientists!) but just that proteins are bloody complicated. If this was any other field besides protein folding, a Tufte disciple like myself would just concoct some clever plot of PrP codons that pits “distance from cysteine” against “age of disease onset” and poof, everything would be illuminated (at least that’s how the magic always seems to work in Tufte’s books!) But alas, a change anywhere in a protein can induce cascading conformational changes throughout the molecule, so just because the F198S mutation isn’t adjacent to 179 or 214 doesn’t mean it couldn’t seriously destabilize the disulfide bond (and for what it’s worth, F and S are very different amino acids).
So, signing off, no conclusions, just some interesting things to think about. Walker et al’s work in PNAS dates from 2001 and so far I haven’t been able to find any followup on it since then, so I’m curious what the current thinking is on the intermolecular disulfide bond idea.