Cell Biology 02: The Plasma Membrane
This semester I’m taking a Cell Biology class at Harvard Extension. My plan is to turn my notes from each lecture into a blog post, figuring out how everything we learned relates to PrP (and maybe huntingtin) along the way. The first class was just review and syllabus, so I’m starting on lecture 2: the plasma membrane.
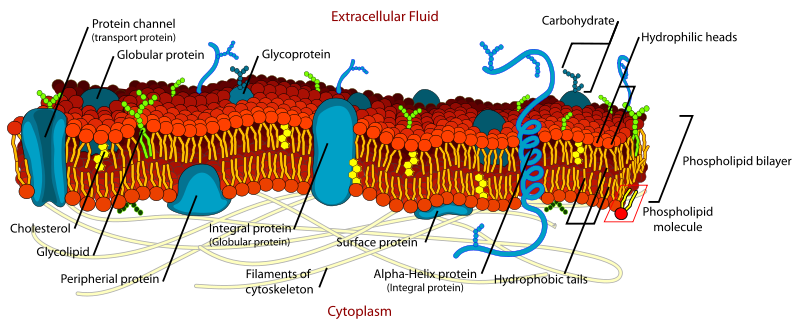
The plasma membrane refers both to the membrane that surrounds cells and also to the membranes that surround organelles within the cell. The functions of the plasma membrane are held to be as follows:
- Compartmentalization (separate organelles from other stuff)
- Scaffolding (give the cell shape)
- Barrier (keep some things out and others in)
- Gatekeeper (do let some things through but not others)
- Monitoring outside signals (receptors on the membrane signal to other proteins inside the cell)
- Energy transduction (transferring energy across the membrane?)
The membrane is made of a lipid bilayer (the red/orange/yellow stuff in the above diagram). It’s made predominantly of phospholipid molecules, which are amphipathic – part hydrophobic and part hydrophilic:

They contain, from top to bottom:
- A polar head group. This can be charged: serine (-), inositol (-), or it can be neutral, with both positively and negatively charged groups: choline, ethanolamine.
- A glycerol linker.
- A phosphate group.
- Two fatty acid chains.
The polar head group faces outward into the aqueous intracellular or extracellular space, while the hydrophobic interior of the membrane is the fatty acid chains. The phospholipids are named after their head groups, thus phosphatidylserine (PS), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI). Then, randomly, there is also sphingomyelin which has an ethanolamine head group but differs from the others in that it has sphingosine (a long carbon chain amino alcohol) instead of one of the fatty acid chains, and an amide linker instead of glycerol.
The fatty parts – the lipids – make up about 50% of the total mass of the membrane. The fatty acid chains can be either saturated or unsaturated, which accounts for some variety in membrane properties. The saturated chains have no double bonds (all the carbons’ bonding capacities are used up by hydrogens), so the chains lie straight, pack tightly and interact much with each other. The unsaturated chains form kinks due to double bonds – most fatty acids in nature are cis fatty acids, meaning all the hydrogens are on one side, so the chain kinks; trans fats, which are abundant in fast food but rare in nature, lie more straight like saturated fats. The kinks in unsaturated cis fatty acids makes them pack less tightly, and the greater amount of space makes for a more ‘fluid’ membrane. Here, ‘fluid’ means it is easier for other things (like transmembrane proteins) to move in two dimensions along the plane of the membrane.
Phospholipids are just most – not all – of the lipid bilayer. There are also glycolipids, which have a glycerol (which is a sugar) instead of phosphate. These make up 2-10% of membrane mass, and are more abundant in the nervous system than elsewhere. Cholesterol is also an important part of the membrane. Here’s what cholesterol looks like:

The OH makes it polar on that end, while the rings and carbon chain on the other end is hydrophobic. So this is also an amphipathic molecule, though it can’t form lipid bilayers on its own. Cholesterol is a steroid, which apparently is a chemical designation referring to that four-ring core, and not a functional designation. Cholesterol is found mostly in eukaryotes and not in bacterial membranes. Its bulky four-ring structure makes the membrane less flexible, the OH head group interacts strongly with other phospholipid head groups thus holding them in place, and the hydrocarbon chain interacts strongly with the phospholipids’ fatty acid chains, making them pack more tightly. For all of these reasons, cholesterol reduces membrane fluidity, though in a temperature-dependent matter (higher temperature means more fluidity).
The two halves of the lipid bilayer are called leaflets: the cytoplasmic leaflet faces (predictably) towards the cytoplasm while the exoplasmic leaflet faces outside the cell or into an organelle. Here is a quick comparison of composition:
- phosphatidylserine (PS) (-): almost all cytoplasmic
- phosphatidylcholine (PC) (neutral) found on both sides, esp. exoplasmic
- phosphatidylethanolamine (PE) (neutral) found on both sides
- posphatidylinositol (PI) (-) almost all cytoplasmic
- sphingomyelin mostly exoplasmic
- cholesterol equal on both leaflets
The exoplasmic side tends to be less fluid. But both sides are pretty fluid, with the phospholipids constantly skating around in 2 dimensions (‘lateral diffusion’), rotating, bending tails (‘flection’), and very occasionally, switching from one leaflet to the other (‘flip-flopping’).
A neat laboratory protocol for studying the fluidity of the membrane is fluorescence recovery after photobleaching (FRAP). In this protocol, a molecule of interest is fluorescently tagged – for instance, a membrane protein is tagged with a fluorescently labeled antibody or fused with green fluorescent protein, or an amphipathic molecule is tagged with a fluorophore. Then you ‘bleach’ part of the membrane with a laser which exhausts the fluorescent properties of the fluorescent molecule in that patch. Then you watch as still-fluorescent molecules from elsewhere in the membrane diffuse into the bleached patch. The main goal here is to see how ‘mobile’ the protein you’re studying is. Generally, membrane proteins are less mobile than the phospholipids because they are so much larger. And if the protein is anchored to the cytoskeleton, it won’t move at all, so the bleached patch will recover very slowly if at all. To control for the effects of membrane fluidity you can also tag the phospholipids themselves and compare the mobility of the protein of interest to that of the phospholipids.
There are two kinds of membrane proteins: integral (top) and peripheral (below). Images thanks to Wikimedia Commons user Foobar:
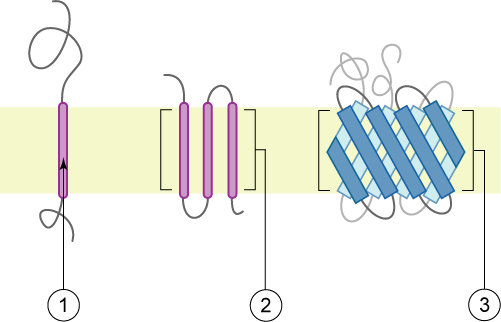
An integral membrane protein (above) has one or more hydrophobic domains anchored in the membrane by simple thermodynamics while the hydrophobic parts stick out into the cytoplasm or extracellular (or intraorganelle) environment). The transmembrane parts might be alpha helices (1 and 2) or beta sheets (3).

The peripheral membrane proteins (above) may have just a segment that buries in the membrane but doesn’t cross it (1, amphipathic alpha helix; 2, hydrophobic loop; 4, hydrophobic interactions with polar head groups) or be post-translationally modified to add an anchor which buries in the membrane (3) or interact with a transmembrane protein (not pictured). The post-translational modifications might attach a fatty acid chain, oligosaccharide, sugars or farnesyl alcohol group (more common on the cytoplasmic leaflet).
All the different molecules that make up the plasma membrane are not evenly distributed across the whole 2-dimensional surface. Instead, there are patches that are more ordered and tightly packed and have 3-5x the concentration of cholesterol and about 1.5x the concentration of sphingomyelin. These patches are called lipid rafts. In the below image, section 1 is normal plasma membrane and section 2 is a lipid raft:

Lipid rafts have all sorts of highly specialized functions and some particular proteins tend to locate there. People who study membrane proteins often add detergent to their solution to solubilize the glycolipids and bring the proteins into solution. That works less well if the protein of interest is found in lipid rafts, which are harder to dissolve.
The membrane is permeable to gases and small uncharged molecules, which can simply diffuse right through according to their own driving force. Naturally, water, ions, small polar molecules and large, uncharged-but-polar molecules are unable to pass through the membrane on their own. Moving these sorts of things requires some form of transport mediated by membrane proteins: either active (using ATP to fight the transported molecules’ own thermodynamic desires) or passive (allowing molecules to follow their own electrochemical gradient).
Passive transport can take a few different forms. First, it can involve channels that allow ions or molecules to diffuse through. These channels might be highly selective or not at all selective, and might be electrically or chemically gated so they only open under certain conditions. Second, it can involve ‘passive transporters’ (aka ‘porters’), proteins which, upon encountering their substrate ion or molecule, undergo a change in conformation which propels that substrate through to the other side. Predictably, they tend to be more specific than channels, but surprisingly (to me at least), the porters are often faster than just straight-up diffusion would be. Porters are divided into three types:

Uniporters just transport one type of molecule, symporters move two molecules in the same direction and antiporters move two molecules in opposite directions. The trick of the symporters and antiporters is that they allow one of their two substrates to move with its electrochemical gradient, and they harness the energy of that molecule’s thermodynamically favorable movement in order to propel the other molecule against its electrochemical gradient. The symporters and antiporters all require that the multiple substrates bind at the same time – they can’t store energy for later.
GLUT1 is the classic example of a uniporter. It imports glucose into cells when extracellular concentrations are higher then intracellular. It is highly specific for glucose, having limited affinity even for mannose and galactose which differ from glucose in just one carbon atom. Once inside the cell, glucose is immediately modified with a phosphate group so it can’t find its way back out. Red blood cells have tons of GLUT1 because, lacking mitochondria, they rely on anaerobic glycolysis for energy, which is really inefficient (only 2 ATP per glucose as opposed to 36 ATP per glucose via aerobic metabolism).
People who study uniporters use antibodies to pull them down, genetically introduce them into cell types where they are not normally expressed (e.g. GLUT1 in skin cells) or mutate their substrate binding region to see how that changes their function. In spite of this, much is unknown – according to Wikipedia we still don’t know the structure of GLUT1 or how it binds glucose.
A classic antiporter is the Na+/Ca2+ exchanger (genes: SLC8A1, SLC8A2, SLC8A3). An alternate term for it, ‘sodium-linked calcium antiporter’ tells you more about its energetics: it’s powered by sodium in order to move calcium against its gradient. Specifically, it accepts 3 Na+ into the cell in exchange for expelling 1 Ca2+. Its affinity to calcium is low enough that it only works at really high calcium concentrations, so it’s useful for preventing excitotoxicity in neurons – getting rid of the excess calcium after repeated stimulus. It’s also found in cardiac muscle cells.
Another classic antiporter is the sodium-hydrogen antiporter (genes: SLC9A1,2,3,5,6,8), which accepts 1 Na+ into the cell in order to export 1 H+ ion. This maintains the preferred pH of 7.2 inside cells. It also is one step in exporting acid into the stomach, because H+ is produced in cells but secreted into the stomach. (But – I had to check – this antiporter is not the target of proton pump inhibitors like omeprazole - those target an active transporter, H+/K+ ATPase).
A classic symporter is the sodium glucose symporter (genes: SLC5A1, SLC5A2 & SLC5A4). Remember how GLUT1 uniporter brings glucose into the cell when its extracellular concentration is higher? When the intracellular glucose concentration is higher than extracellular and cells need to import glucose against its concentration gradient, this symporter does so by accepting 2 sodiums into the cell in order to bring one glucose in.
Active transport relies on pumps that couple ATP hydrolysis with movement. You may have noticed that sodium seems to be the common currency of the exampels of symporters and antiporters listed above. They all allow sodium to move into the cell, as is thermodynamically favorable, in order to export something the cell doesn’t want. Predictably, someone has to do the hard work of maintaining that sodium gradient while everyone else is mooching off of it. That someone is Na+/K+-ATPase, which expends 1 ATP in order to export 3 Na+ and import 2 K+, both against their respective concentration gradients. This thing is a workhorse, accounting for 20% of energy consumption in most animal cells and 67% in neurons. You don’t get anything for free, so if it just doesn’t feel right to even call the symporters and antiporters ‘passive’ transporters, you can call them by their other name, which is ‘secondary active transporters‘, in recognition of the fact that, while they don’t burn ATP directly, they do use energy by relying on actively maintained sodium gradients.
The active transporters are divided in to F, P, V, and ABC families.
P refers to P-type ATPases which have two catalytic alpha domains (one of which is phosphorylated hence the ‘P’ name) that bind and hydrolyze ATP, and two regulatory beta domains. These include the Na+/K+-ATPase mentioned above as well as SERCA calcium pumps, which stores calcium in the sarcoplasmic reticulum, and the H+/K+ ATPase which acidifies your stomach, mentioned above.
ABC refers to the ATP-binding cassette family of hundreds of proteins. They have two transmembrane domains and two ATP-binding domains. Some of them move molecules and not just ions. This class is said to include CFTR, the protein whose loss of function is responsible for cystic fibrosis. But CFTR is a bit weird and, though it evolved from the active transporters, it does not actually pump anything uphill against a concentration gradient. Rather, it allows passive diffusion of chloride and thiocyanate ions along their gradients, but only when opened by an ATP-powered gate. Loss of function mutations in CFTR make it impossible for these ions to diffuse, which in turn eliminates the thermodynamic incentive for water to osmose out of the cell. Without that extra water from osmosis, the extracellular mucus becomes too thick, leading to coughing and chest infections, the hallmarks of cystic fibrosis.
Another important ABC family transporter is MDR1 (gene: ABCB1). MDR stands for multi-drug resistance. This amazing protein found in animal cells is not very specific but somehow can recognize a broad range of ‘xenobiotic‘ molecules – i.e. foreign compounds that we don’t produce and don’t normally have in our cells – and, using energy from ATP, kick them out of the cell. Presumably this keeps the cells from accumulating toxins from the environment. But it also makes treatment difficult because it exports some antiretroviral drugs and chemo drugs – the latter especially in tumor cells that acquire mutations that heighten the action of MDR1. In this way, tumors can sort of evolve resistance to cancer drugs. This is especially a big problem in liver cancers. An old, boring theory about MDR1 was that it was just an ATP-powered channel, but FRAP-like experiments have now shown that it’s in fact something much more interesting: a flipase. That means that rather than just shooting the molecule out of the cell, MDR1 binds the molecule and then flips a section of the membrane, swapping the exoplasmic and cytoplasmic leaflets. It’s like punching a door in a fence in Super Mario World:
The F family are found in bacteria, mitochondria and chloroplasts, and based on their Wikipedia description, don’t sound like active transporters at all.
The V family are found in many other organelles and are responsible mostly for pH maintenance. Lysosomes - one place where misfolded proteins and other cellular waste go to get broken down – have pH 4.8 (compared to 7.2 in the cytosol), which is a clever evolutionary trick. The lysosomes are full of enzymes that could break down everything in the cytosol, which is dangerous, but the enzymes evolved to only function at low pH, so if the lysosome ever ruptures, the enzymes won’t destroy the cell because they won’t function in the cytosol. This pH gradient between lysosome and cytosol is maintained by V ATPases.
PrP and the plasma membrane
PrP in humans is 253 codons long but the final protein is only 208 amino acids. 22 N terminal and 23 C terminal amino acids are signal peptides which direct the protein into the endoplasmic reticulum, where it gets post-translationally modified. That modification cleaves off the signal peptides, replacing the C-terminal segment with a glycosylphosphatidylinositol (GPI) anchor (attached to 231S) which makes PrP technically a peripheral membrane protein (though GPI-anchored proteins have some properties more like integral membrane proteins), with the GPI buried in the exoplasmic leaflet of the plasma membrane and the protein sticking out into the extracellular space. GPI-anchored proteins tend to congregate in lipid rafts. PrP is also a glycoprotein because during post-translational modification it gets glycosylated (has carbohydrate side chains added) at amino acids 181N and 197N.
There has been some debate over whether PrP’s anchoring to the membrane via the GPI anchor is important in prion disease. Experiments seem to suggest that when PrP is not GPI-anchored, it can still misfold but tends not to do so on a large scale: GPI anchorless mice are tough to infect with prions [Klingeborn 2011] and prions don’t propagate well in vitro when the PrP is not GPI-anchored [Priola 2009]. PrP’s localization in lipid rafts and proximity to cholesterol are thought to be somehow important for conversion to PrPSc, which might be part of the explanation for the (albeit limited) therapeutic efficacy of statins and amphotericin B in prion-infected mice. Some people think the N terminus of PrP interacts with another lipid raft protein (possibly Glypican-1 [Taylor 2009]) and that this interaction is necessary (or at least pretty important) for conversion to PrPSc.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.