Cell Biology 06: The Cytoskeleton Part II: Tubulin
These are notes from lecture 6 of Harvard Extension’s Cell Biology course.
Last week we covered microfilaments, which are made of actin. This week: microtubules, made of tubulin. Why, you ask, does the cytoskeleton need two separate systems? Consider that microfilaments are just chains of individual subunits, whereas microtubules are literally tubes (as we’ll see shortly) – hollow cylinders with walls made of chains made of dimers. These different structures mean different capabilities: microfilaments can more easily form spontaneously, and can branch (with the help of Arp2/3 and other complexes discussed last time) in order to form different network shapes and connect different parts of the cell. Microtubules rely more heavily on nucleation in order to form, and are basically a spoke network connecting the centrosome to the cell’s periphery. In practice, microfilaments are abundant in the cell cortex and heavily involved in contractile movements and cell motility, while microtubules are most involved in organizing organelles and serving as tracks for anterograde and retrograde transport.
Microtubules
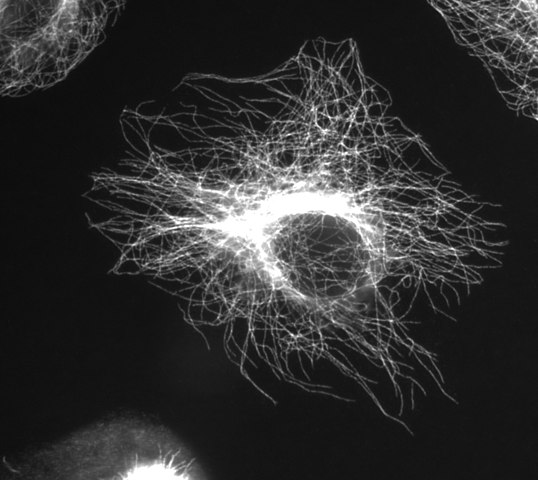
(Image thanks to Wikimedia Commons user Jeffrey81)
In addition to Wikipedia, previous editions of the Lodish and Cooper cell biology textbooks available on NCBI are also excellent references.
The tubular structure of microtubules is sturdier than microfilaments, allowing a lot of heavy duty pulling and pushing. Though sturdy, microtubules are temperature-dependent – they depolymerize if cooled to 4°C and will repolymerize again if heated to 37°C provided that GTP is available.
The basic building block of microtubules is an α-tubuilin/β-tubulin dimer (encoded by TUBA_ genes and TUBB_ genes respectively). The individual tubulin proteins each weigh ~55kDa, so at ~110Da / amino acid that’s on the order of 500 amino acids. Both α- and β-tubulin bind GTP, but α pretty much just holds onto it forever, whereas in β it can be exchanged or hydrolyzed to GDP and then exchanged for new GTP again.
Strands of consecutive α/β dimers make up protofilaments, and 13 protofilaments arranged side-by-side into a cylinder make a microtubule. The space in between the protofilaments is called a ‘seam’. The whole microtubule is ~25 nm in diameter. You can kind of see its structure (made of dimers and protofilaments) in the Inner Life of the Cell segment about microtubules:
Within each dimer, the β subunit is the (+) end, favored for polymerization and the α is the (-) end, favored for depolymerization. Microtubules form in basically the same three steps as microfilaments – nucleation, elongation and steady state. But unlike microfilaments they do not nucleate easily on their own, so nucleation requires microtubule organizing centers (MTOCs). Non-dividing cells each have but one MTOC, called the centrosome. (This is also depicted in the video above). Located near the nucleus, the centrosome is hub to a radial configuation of microtubules with (-) ends pointed in and (+) ends pointing to the cell periphery.
The centrosome is composed of 2 cylinders called centrioles. Each centriole consists of each 9 sets of 3 laterally fused microtubules, surrounded by an amorphous ‘pericentriolar material‘ rich in things that promote nucleation – particularly γ-tubulin ring complexes (gamma tubulin is encoded by the TUBG_ genes). γ-tubulin is thought of as a ‘split washer’:

The model is that the split ends allow γ-tubulin to bind to α-tubulin – i.e. the (-) end of a to-be-formed microtubule – providing the nucleation seed for a microtubule to form.
Microtubules can be formed in vitro. The dynamics depend mostly on critical concentrations. The (-) end is less inclined towards polymerization than the (+) end, so it has a higher critical concentration. If the actual concentration is between the two ends’ critical concentrations, treadmilling does occur.
When a microtubule suddenly starts to dissociate this is called a ‘catastrophe’. Catastrophes have some interesting energetic dynamics. Recall from earlier that beta-tubulin, which is the (+) end where elongation happens, can be either GTP- or GDP-bound. It is a more stable part of its microtubule when it is GTP-bound; GDP-bound tubulin is apt to dissociate. Microtubules form primarily by addition of GTP-bound beta-tubulin at the (+) end, but after being added, the beta-tubulin molecules later hydrolyze their GTP, leaving them GDP-bound. So there is a sort of ‘tip’ of the microtubule that is GTP-bound, while beta-tubulin deeper down the microtubule, added longer ago, are GDP-bound. If the frontier of GTP hydrolysis catches up to the tip, or if something happens to sever the microtubule, then the less stable, GDP-bound beta-tubulin becomes exposed, and the protofilaments will start to peel away like “ram’s horns”. If this happens, the microtubule will disassemble until it hits an “island” of GTP-bound beta-tubulin somewhere further down the straw. Catastrophe can be ‘rescued’ by the addition of new GTP-bound tubulin dimers that will cap the ends and stabilize the protofilaments, allowing the microtubule to re-form. You can see in this video that depolymerization of microtubules can be much faster than polymerization:
Here are some compounds useful for studying microtubules. Colchicine, an anti-gout drug, binds free alpha-beta dimers, reducing their supply for microtubule formation and thus promoting depolymerization. Nocodazole also interferes with new microtubule formation, and because formation of new microtubules is important for mitosis, nocodazole is an antineoplastic cancer drug. Conversely, paclitaxel (taxol), another anti-cancer drug, works by stabilizing microtubules, since breakdown of existing microtubules is also important for mitosis.
Microtubule-associated proteins (MAPs) have various roles. Noteworthy are MAP4 (in non-neuronal cells), MAP2 (in neurons) and Tau (in neurons; coded by MAPT gene). All of these have in common that they act to stabilize microtubules, shifting the kinetics in favor of microtubule growth and against catastrophe. Each contains an 18 amino acid stretch with positively charged amino acids that bind to the negative part of microtubules. They may act to bundle multiple microtubules together [Dehmelt & Halpain 2005], with MAP2 holding the microtubules at a greater distance due to its longer arm (compared to Tau).
The MAPs are regulated by phosphorylation: the MARK [Drewes 1997] protein kinases covalently bind phosphate groups to S, T or Y amino acids in the MAP proteins, reducing their ability to bind microtubules. (Cyclin dependent kinases also regulate the MAPs during mitosis). It is thought that these MAPs act to determine the physical structure of cells. In neurons, MAP2 is found in dendrites and Tau is mostly in the axon. Mutations in the MAPT gene that codes for Tau cause frontotemporal dementia (FTD). Hyperphosphorylated Tau is found (though we don’t know why) in Alzheimer’s patients’ brains. Mouse models of both of these diseases show axonal degeneration [Gotz & Ittner 2008 (ft)], consistent with Tau not being able to do its job of stabilizing microtubules in the axon.
Another class of proteins, named +Tips for their binding to the (+) end of microtubules, can protect against catastrophe. They appear to extend a fair ways down into the microtubule. This video shows EB-1 being pushed outward because the ends of microtubules are growing:
Other end-binding proteins promote catastrophe rather than stability. Kinesin-13 acts to curve the end of the protofilaments, lowering the threshold for catastrophe. Stathmin (STMN1 gene; aka Op18, where Op stands for oncoprotein) binds to tubulin dimers within a protofilament, promoting both immediate catastrophe and possibly also GTP hydrolysis, which by removing “islands” of GTP-bound beta-tubulin would represent more of a long-term investment in favor of catastrophe. Stathmin is regulated by phosphorylation. Katanin (after the Japanese sword katana) literally severs microtubules.
Microtubule motor proteins are largely similar to microfilament motor proteins. They come in two families: kinesins, most of which move anterograde i.e. toward the (+) end; and dyneins, most of which move retrograde i.e. toward the (-) end. They ‘walk’ like this:
Kinesin and dynein are involved in moving organelles, endocytosis and exocytosis, and chromosome segregation during meiosis & mitosis.
Kinesin-1, the best studied of the kinesins, is a ‘conventional’ kinesin in that it operates as a tetramer composed of two heavy chain units (KIF1 genes e.g. KIF1A) that comprise the ‘head’ domains that hydrolyze ATP and bind the microtubules, and two light chain units (KLC genes e.g. KLC1) that comprise a ‘tail’ that binds the cargo, in this case vesicles. A linker domain (which protein is this part of?) allows for dimerization of two heavy chains.
Here’s a video of it in action. Note that the video calls it a dimer instead of a tetramer; I think that’s because they are only considering the heads and not discussing the tails.
Some of the other kinesins differ a bit:
- Kinesin-2, which also does vesicle & organelle transport, is a heterotrimer with two different (though related) heavy chains and another polypeptide it uses to regulate its cargo.
- Kinesin-5 has heads at both ends instead of a head and tail, so that it walks in opposing directions on two different microtubules, pulling them together. This is called ‘bipolar movement’.
- Kinesin-14 is the only known kinesin that moves towards the (-) end, and it is involved in mitosis.
The principles of the ‘walking’ movement are pretty much the same for all of the kinesins, though, and are what’s depicted in the last video above. When not bound to a microtubule, the ‘heads’ are both ADP-bound. One head will happen to encounter a microtubule and bind to it, releasing its ADP, allowing an ATP to replace it. The ATP binding induces conformational change that pulls on the linker, swinging the other, ‘lagging’ head forward into leading position where it binds to the microtubule. The original head then hydrolyzes ATP which releases phosphate (which is abbreviated as Pi in the video) and provides the energy for the one energetically uphill step in this process, which is breaking away from the microtubule.
Myosin (the motor protein that walks on microfilaments) and kinesin have very similar structures but no amino acid sequence similarity. Thus they are believed not to be paralogs, but rather an example of convergent evolution.
People talk a lot about kinesins in part because we don’t understand dyneins quite as well. Dyneins are huge proteins, made of 2 large, 2 intermediate and 2 small subunits. Their sheer size has made them difficult to isolate and characterize and their workings are not well understood. We do know that the dynactin complex (a multi-protein complex; the dynactins themselves are the DTCN_ genes) is involved as an ‘adapter’ that links dynein to cargo.
This video summarizes the whole cytoskeleton and ropes in much of the material from last week and this lecture:
PrP
As mentioned in the secretory pathway notes, PrP is GPI-anchored to the membrane and undergoes endocytosis very regularly, creating endocytic vesicles with PrP in them. Encalada 2011 finds that Kinesin-1C and DHC1 (dynein heavy chain 1) are responsible for transporting these PrP vesicles anterograde and retrograde respectively. When Encalada knocked out, knocked down or inhibited Kinesin-1C, anterograde and retrograde movement were reduced, and similarly when DHC1 was disrupted. So it seems that these two complexes, though they move in opposing directions, activate one another. And interestingly, disrupting these motor proteins didn’t prevent PrP vesicles from associating with motors – they just didn’t move as fast or as often. The paper has a number of other cell biology conclusions about the nature of how vesicles activate motor proteins and how the direction of transport is determined.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.