How to, and how not to, knock out PRNP
About a year ago, I sat down to get my facts straight on zinc finger nucleases (ZFNs) and TAL effector nucleases (TALENs), two genome-editing technologies. But even as I wrote those words, a newer technology called CRISPR/Cas9 was beginning to make waves. In the past few months, CRISPR has become big news, especially after Editas Medicine here in Cambridge raised $43M to develop human therapeutics based on CRISPR genome editing.
Perhaps CRISPR’s biggest advantage over ZFNs and TALENs is its size: it uses RNA, rather than peptides, to recognize its target site in the genome. The smaller size of the DNA required to code for the RNA plus the Cas9 nuclease makes it easier to, say, clone into your viral vector of choice. It also appears to be highly efficient: for example, the landmark paper on editing human cell lines with CRISPR/Cas9 achieved non-homologous end-joining in 2-4% of clones in iPS and as many as 25% of clones in other cell lines, and successful homologous recombination in 3-8% of clones [Mali 2013]. My, how efficiency has improved since the bad old days: when the first PrP knockout mouse was created using plain homologous recombination (with no way to create double strand breaks to speed the process up), only 1 in 5000 clones had the knockout allele, even after selecting for G418 resistance [Bueler 1992]. With CRISPR you don’t even need drug resistance cassettes.
To be sure, CRISPR/Cas9 does not, in and of itself, solve the problem of safe and effective delivery for use in vivo. Getting it into the brain, and to a lesser extent other tissues of interest, will continue to be a challenge. And even in a relatively low-immunogenic viral vector like AAV9, there is still the question of whether people’s immune systems will recognize the Cas9 nuclease as the non-self protein that it is. These sorts of issues are the reason why the first clinical trial of genome editing with ZFNs used an ex vivo approach, deleting CCR5 in people’s T cells and then transplanting them back in [Tebas 2014].
Still, the potential of CRISPR/Cas9 is enough to raise the question of what could be done with it therapeutically if we could get it into the brain. When people talk about “genome editing”, non-scientists often imagine this means correcting a missense mutation in someone’s genome. But even with directed double-strand breaks, non-homologous end joining occurs at a much higher rate than homologous recombination, so a knock-in therapeutic strategy in vivo doesn’t seem very likely to me (though perhaps it could be done ex vivo by selecting cells before transplanting them back in). A much easier target would be a gene where the goal is deletion, not correction.
In that sense, prion diseases are an ideal target for CRISPR/Cas9: a class of rapidly lethal gain-of-function disorders that depend upon expression of a single gene whose knockout appears well-tolerated. As a way to deplete PrP and slow or stop prion diseases, knocking out PRNP with CRISPR/Cas9 would have several advantages over RNAi or ASOs. It would outright eliminate, not merely reduce, PrP production in cells where you manage to hit both alleles, and sustained therapeutic benefit wouldn’t depend on sustained expression of the viral vector. Even transient expression of the CRISPR/Cas9 construct would be enough to permanently knock out the gene, so that if the viral vector were later silenced the (beneficial) damage would already be done.
This got me thinking, if we could use a viral vector to deliver CRISPR/Cas9 to delete PRNP in the brain, where exactly would we want to cut?
PRNP is an exceptionally pathogenic gene: not only do missense mutations cause prion disease, but even the wild-type sequence can cause spontaneous disease if the protein is overexpressed [Westaway 1994], expressed ectopically under a foreign promoter [Tremblay 1998], secreted instead of GPI-anchored [Stohr 2011], or interstitially deleted [Shmerling 1998, Li 2007]. So when you cut, cut carefully.
There exist several lines of PrP knockout mice, so I looked back to the original papers to see how people targeted Prnp in the mouse embryo 20 years ago. Mouse Prnp has three exons, and the entire coding sequence is in exon 3, as shown below.
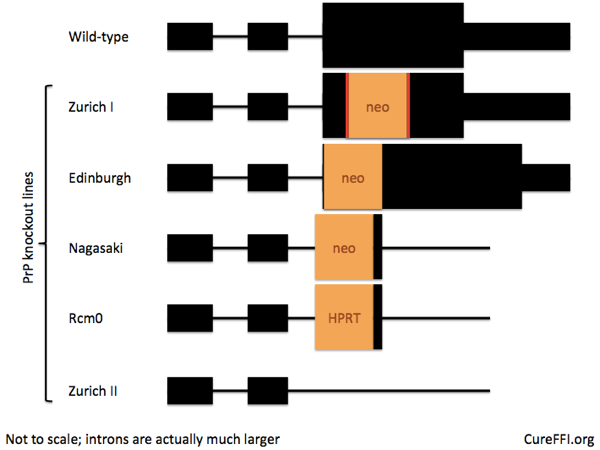
The first knockout was the Zurich I line [Bueler 1992], which has a stop codon, neomycin resistance cassette, and then two more stop codons, inserted into Prnp‘s only coding exon in place of codons 4-176. These mice do express PrP mRNA, it just doesn’t code for a viable version of PrP anymore. These mice were quickly adopted by the Prusiner lab [Prusiner 1993] and are the most widely used PrP knockout line today.
Next came the Edinburgh line [Manson 1994], which inserts a neo cassette right at the beginning of exon 3 after the splice acceptor site. The creators found that the knockout allele led to a complete loss of PrP mRNA expression.
Third came the Nagasaki line [Sakaguchi 1995], and with it a lesson for the ages [Sakaguchi 1996]. I told a bit of this story in the pi post, but it merits a quick re-telling here. Whereas the Zurich and Edinburgh lines had both been reported as developmentally normal, the Nagasaki line had a late (~70 weeks of age) onset ataxia caused by neurodegeneration in the Purkinje cells of the cerebellum.
When the Nagasaki line’s phenotype was first published, it was big news. By that time, the only PrP knockout phenotypes anyone had found were an electrophysiological difference observed in hippocampal slices [Collinge 1994] and a mild change in circadian rhythms [Tobler 1996]. A clear – and clearly deleterious – in vivo phenotype earned the Nagasaki line its place in Nature. Not only did this supposed knockout phenotype point to a native function for PrP, it also raised the question of whether loss of function might be at least partly responsible for the neurodegeneration caused by prions.
But this phenotype was bound to prove contentious: after all, the Zurich line had been monitored out to 90 weeks and had no such phenotype. Sakaguchi et al ventured a few possible explanations. Perhaps a mouse genetic background effect controlled the age of onset of the ataxia, or perhaps the Zurich knockout construct was able to retain some of PrP’s native function. Notably, the authors had omitted the classic rescue experiment in which one checks whether a transgene can reverse the knockout phenotype. So any real sticklers watching this saga probably thought that the late-onset ataxic phenotype was an artifact of the genetic manipulation, soon to be disproven by a failed rescue experiment.
But the sticklers were in for a much more interesting show than expected! It took the Nagasaki line’s creators three years to complete and publish the rescue experiment, but it worked: a PrP transgene did indeed prevent the late-onset ataxia [Nishida 1999]. At almost the exact same time, though, another group found that the Nagasaki mice expressed a heretofore unknown gene in their brains [Moore 1999]. This gene turned out to be a paralog of PrP itself, and located 16kb downstream of Prnp, so the second group christened their new protein Doppel, for downstream prion protein like. (Many later authors, including me in an earlier revision of this blog post, mistook it as standing for doppelgänger). Dpl, as it was known for short, was normally expressed during embryogenesis but not in the adult brain – except in the Nagasaki mice.
This new gene’s ectopic expression in the brain appeared, then, to explain the phenotype of the Nagasaki line, despite the successful rescue experiment. Curiously, Prusiner was a co-author on both the transgene rescue paper and on the Doppel paper – one has to wonder what he thought about the whole saga. People later generated two additional PrP knockout lines (Rcm0 and Zurich II) with the exon 3 splice acceptor deleted, and confirmed that ectopic CNS expression of Doppel indeed causes neurodegeneration which can be rescued by co-expression of PrP [Li 2000, Silverman 2000, Rossi 2001, Moore 2001].
The mechanism for the Doppel-induced neurodegeneration and the rescue by PrP remains unknown to this day (though there are some interesting theories discussed here). However, the details of how Doppel (gene symbol Prnd) came to be expressed in the brain are now clear, and are perhaps best explained [Weissman & Flechsig 2003 (ft)], particularly in Figure 1C. Absent the Prnp exon 3 splice acceptor site, the Prnp transcript reads straight through the end of the Prnp gene, splices to two additional exons in the 16 kb between Prnp and Prnd and then splices into all of the Prnd exons. The end result is a chimeric transcript under the control of the prion promoter and thus expressed in the brain, but with Doppel in its protein-coding sequence. This chimeric transcript even made its way into RefSeq and can be seen in the UCSC Genome Browser. The two genes are separated by 16 kb (first line) but can be fused with an extra exon in between (second line):
{kind=link}

In humans, PRNP only has two exons, and PRND slightly further away, 20kb downstream. It’s not known whether a similar chimeric transcript could be created by deleting the PRNP exon 2 splice acceptor site, but it’s certainly a risk one would want be aware of and test out in cell lines before going in vivo with CRISPR/Cas9.
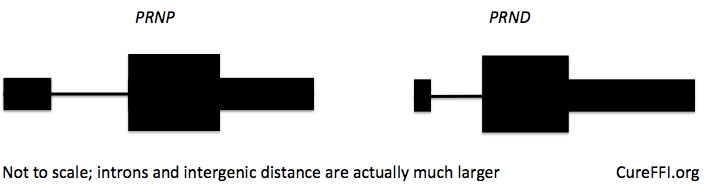
Indeed, probably any CRISPR/Cas9-based strategy would need to be tested to figure out its effects on expression of PRNP and neighboring genes. For almost any part of the gene you can think to target, it is at least possible to imagine a potential bad outcome, as diagrammed below. We have no evidence that any of these things would actually occur, but still, any strategy would need validation in neuronal cell lines to see what happens.

Depending where one targets PRNP with CRISPR/Cas9 (left), it will be important to double check that the potential hazards (right) do not occur. Here is a little more explanation, from left to right:
- Overexpression [Westaway 1994], and ectopic expression [Tremblay 1998] can both be toxic. Neither of these things seems particularly likely to occur if one is trying to delete the prion promoter, but it will be important to check to be sure. Of course, to deplete PrP and stop prion disease, the most important thing will just be to see that PrP expression is abolished entirely.
- It’s not known what would happen if exon 1 and/or its splice donor were deleted. Would the whole 10kb intron function as a giant 3′UTR and would the coding sequence still be translated? Probably not, but we’d need to do the experiment to find out.
- Deleting the exon 2 splice acceptor might create a neurotoxic PRND fusion transcript, or this problem might only occur in mouse and not in human.
- Disrupting the N terminus could create an interstitial deletion mutant which is neurotoxic [Shmerling 1998, Li 2007]. This doesn’t occur if the very N-terminal tip is deleted, but N-terminal deletion mutants are still competent of creating prions if the deletion extends up to the equivalent of human codon 90 [Fischer 1996]. Prion susceptibility is apparently abolished if the deletion goes up to codon 106 [Weissman & Flechsig 2003 (ft)]
- Disrupting the C terminus might create a secreted frameshift mutant similar to D178fs, which was recently reported in a patient with prion disease [Matsuzono 2013]. The most N-terminal stop codon associated with prion disease in humans is Y145X [Ghetti 1996], and a very early stop codon (homologous to human G29X) appears tolerated in goats [Benestad 2012], so deletions more N-terminal might be fine, but then see the next bullet point above this one.
Luckily, according to the CRISPR FAQ, it’s pretty easy to find sites to target with CRISPR, so it should be an option to try all of these in a neuronal cell line and have a look at the distribution of outcomes. Some frequency of bad outcomes is tolerable: if targeting the N terminus creates a neurotoxic interstitial deletion mutant 1% of the time but causes a total loss-of-function frameshift mutant 50% of the time, that would still be a clear therapeutic benefit on the net. There may also be a possibility of deleting the whole gene: CRISPR/Cas9 has been used to create deletions as large as 1 Mb [Xiao 2013], though I haven’t yet been able to find any data on what size of deletion gives the highest efficiency. Obviously, to go in vivo, you would want the deletion to occur in the highest possible percentage of cells.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.