Genetics 02: 'Introduction to yeast genetics and complementation'
These are my notes from lecture 02 of Harvard’s Genetics 201 course, delivered by Fred Winston on September 5, 2014.
History and motivation for studying yeast
Louis Pasteur first demonstrated that alcoholic fermentation required a living organism, yeast. O. Winge is considered to be the father of yeast genetics — he figured out that:
- Yeast can exist stably in either a haploid or diploid state
- Yeast have two stable mating types (called a and α, analogous to male and female)
Beer brewing was the original motivation for the study of yeast. It later became popular as a model organism because:
- It is a unicellular eukaryote
- You can use microbial techniques to study it
- A remarkable proportion of its biology is conserved in other eukaryotes including humans
It also has a more tractable genome than we do:
| S. cerevisiae | H. sapiens | |
|---|---|---|
| Chromosomes | 16 | 23 |
| Genome (bp) | 1.2e7 bp | 3e9 bp |
| Genes | 6,600 | ~20,000 |
It is estimated that ≥30% of yeast genes have human orthologs (based on sequence similarity not function).
Yeast is a particularly good system for studying cell division, secretion, chromatin structure, mitochondria, genomics, evolution, complex traits, and bioengineering.
Yeast is generally considered a particularly bad system for studying anything unique to multicellular organisms (for instance, neurobiology, heart development, etc.), though Fred points out that this list has shrunk over time - for example, Susan Lindquist has demonstrated the utility of yeast for studying neurodegeneration.
Mitosis
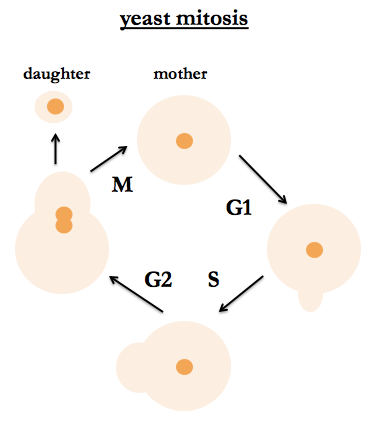
S. cerevisiae are budding yeast. Their mitotic cycle is the same for haploids and diploids. One can tell their position in the cell cycle by bud size, nuclear division (observable by staining DNA) and DNA content. After budding, the mother cell is stained with a bud scar, so it is possible to tell how many times the cell has given birth, and people study aging in yeast and how many times a yeast cell can give birth before it dies. Fred recommends a classic work by Hartwell (I believe he is referring to [Hartwell 1973]).
Meiosis
Yeast have two mating types, a and α. All matings must involve one of each. If you cross an a cell with a genetic marker B and an α cell with genetic marker b, you get a diploid cell with both mating types (a/α) and both markers (B/b). Only diploids can undergo meiosis. In the lab, people induce meiosis by nitrogen starvation. In meiosis, four haploid spores are formed. The result is known as an ascus or tetrad. Two of the four will have B, and two will have b. Just like in Mendel’s work, if a trait segregates 2:2, it is encoded by a single gene. Assuming the B/b-determining gene is not in linkage with the mating type, then on average you’ll get Bα, Ba, bα and ba in a 1:1:1:1 ratio. Mating type is determined by two closely linked genes, whose alleles are named α1/a1 and α2/a2.
See next lecture for a more detailed description of how meiosis works in yeast.
Secretion
Much of yeast secretion was elucidated through a very clever mutant hunt [Novick 1980].
Yeast nomenclature
- A wild-type gene is written in all-caps italics: CIS1
- A mutant gene is written in lowercase italics, sometimes followed by a hyphen and allele number: cis1-1
- Proteins are written in initcaps: Cis1
- Phenotypes are written with initcaps and superscripts: CisR and CisS
How to do a mutant hunt in yeast
There are two possible strategies for isolating mutants in yeast.
-
Mutagenize and screen for mutants. Consider for instance the study of cisplatin, a DNA-binding chemotherapy drug which is both toxic and can give rise to resistance, by Smith et al. They looked for cisplatin-resistant yeast mutants. The trait of cisplatin resistance is denoted CisR. To do this sort of study you need a mutagen; one option is ethyl methanesulfonate (EMS), which causes G→A / C→T transitions (Fred writes this GC→AT which I find confusing). You can mutagenize yeast with EMS and then plate the yeast on petri plates containing cisplatin.
-
Screen the yeast deletion set. There exists a commercially available library of yeast strains each of which has one gene deleted and replaced with a gene encoding resistance to geneticin (aka G418, hence the resistance trait is denoted G418R).
Here is how you delete genes to make such a library. Say you want to delete a hypothetical chromosomal gene we’ll call GEN1. Start with a diploid wild-type cell and a plasmid encoding the G418R phenotype. PCR amplify the G418 resistance gene with primers that have ~40bp of homology to the region flanking GEN1. Transform yeast with the PCR product, and by homologous recombination some cells will end up with the G418 resistance gene replacing one of their copies of GEN1. Then select for the G418R phenotype. Sporulate the resistant yeast and separate tetrads.
The creation of these deletion libraries revealed that ~20% of genes are essential for viability (presumably this reveals itself when you find that only 2/4 of the tetrad offspring are viable, and those 2 are not resistant to G418). The ~80% that are viable can be purchased for ~$2000 in either diploid or haploid forms.
Here are some pros and cons.
The “library” approach lets you immediately know the identity of the mutated gene without sequencing. You also know it’s only one gene, which is simpler.
The “mutagenize” approach allows you to find gain-of-function mutations. It is higher throughput because you don’t need to run thousands separate plates in parallel. You can find epistatic effects, as you might have mutations in >1 gene. You can also find mutations in essential genes (presumably the idea is that there may be viable hypomorphic alleles of these). (Though the “library” people have now responded to this by creating libraries of temperature-sensitive mutants of essential genes).
Suppose you’ve done a “mutagenize” approach and have isolated CisR mutants. The next thing you want to do might be to figure out if the trait is dominant or recessive. Mate each mutant with a wild-type strain. Suppose we found two mutants, cis1 and cis2 and we mate an “a” cell of each with a wild-type cell.
| diploid genotype | phenotype | conclusion |
|---|---|---|
| cis1/+ | CisS | recessive |
| cis2/+ | CisR | dominant |
Only for recessive mutations, you sometimes move on to a complementation test.
| cross | diploid phenotype | complement? | number of genes |
|---|---|---|---|
| cis1 × cis3 | CisS | yes | 2 |
| cis1 × cis4 | CisR | no | 1 |
cis1 and cis3 are said to “complement” each other because each can supply what the other is missing. This means they must be mutations in different genes. (Remember we only do this for recessive mutations, i.e. when the wild-type is dominant to the mutant). cis1 and cis4 do not complement each other, implying they are different mutations in the same gene. Complementation is a test of function, always done by mating two haploids to get a diploid and checking its phenotype.
How can you ever know that you’ve found all of the possible genes in the yeast genome whose mutation can engender cisplatin resistance? If we isolate 100 mutants from our mutagenesis screen and find that none complement each other (i.e. they are all in one complementation group) then we can be fairly confident that there is only one gene responsible for this trait. If we isolate 100 mutants and find 73 complementation groups, then odds are we haven’t found all of them. The number of mutants isolated per gene with potential to confer cisplatin resistance is determined by a Poisson distribution. You can use this distribution to estimate the number of zerotons out there - i.e. the number of genes that could have conferred cisplatin resistance, but for which you happened to find no mutants. However the Poisson distribution is but a crude approximation, because the prior of finding a mutant for any one gene is determined by gene size, the gene’s effect on viability, presence of paralogs which might mask its phenotype, and perhaps other factors (one student suggested chromatin structure).

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.