Genetics 07: 'Cytoplasmic inheritance and complex traits'
These are my notes from lecture 07 of Harvard’s Genetics 201 course, delivered by Fred Winston on September 22, 2014.
Cytoplasmic inheritance
There are at least three types of cytoplasmic inheritance:
- Mitochondria
- Chloroplasts
- Prions
Mitochondria
Yeast mitochondria have an 86kb genome (compare to only 16.6 kb in humans). The mitochondria have their own transcription and translation machinery and tRNAs. In mitochondria, the codon TGA encodes tryptophan instead of stop. Most proteins involved in mitochondria are encoded in the nucleus, though, and arrive at the mitochondria via a signal sequence at their N terminus.
Through glycolysis, glucose is converted into pyruvate. In the absence of oxygen, pyruvate can then be fermented to produce ethanol and 2 ATPs. In the presence of oxygen, respiration and oxidative phosphorylation can produce CO2, H2O and 36 ATPs. Note that domesticated yeast used in beer and wine production have genetic variants that allow them to perform fermentation even in the presence of oxygen. Mammals cannot perform fermentation and, in the absence of oxygen, instead perform anaerobic metabolism that produces lactic acid and 2 ATPs.
In humans, mtDNA mutations are responsible for a number of diseases with neurological, cardiac, respiratory and other phenotypes. S. cerevisiae has proven useful for modeling these mutations, because mtDNA is not essential for viability in yeast, unlike in other eukaryotes. In 1949, Boris Ephrussi discovered yeast mutants without any mitochondrial function. They produced smaller colonies which he called “petites”. To this day, yeast with defects in mitochondrial function are called “petites”, even if the causal mutation is in the nuclear genome. The petite mutants cannot grow on lactate, a carbon source which requries oxidative phosphorylation:
| glucose | lactate | |
|---|---|---|
| wt | + | + |
| petite | + | - |
Petites are divided into several classes:
- ρ0 (rho0) has a complete loss of mtDNA. They still have mitochondria, maintained by nuclear genes, but the entire mitochondrial genome is gone, and the mitochondria are non-functional.
- ρ- (rho-) have partial deletions of mtDNA.
- mit- have point mutations in mtDNA.
- pet have mutations in nuclear genes that are necessary for mitochondrial function.
Petite mutants can be categorized by performing petite × wt crosses:
| cross | Lac+:Lac- |
|---|---|
| pet484 × wt | 2:2 |
| *ρ0 × wt | 4:0 |
In other words, mtDNA loss-of-function mutations can be rescued by crossing with wild-type, which repopulates all the progeny with healthy mitochondria.
Complex traits
In the lab it is considered most desirable to find Mendelian, single-locus mutations where you get 2:2 segregation and it is easy to find a causal gene. In reality most traits are complex, with contributions from many loci.
Let’s say for example that our CisR mutants are resistant to 100 μg/mL of cisplatin. Consider our earlier cis1 mutant, which was isolated from a mutagenesis screen and proved to be a single locus Mendelian mutation. If we perform a cis1 × wt cross, we will find this:
set.seed(1)
cisr = sample(c(0,100),size=100,replace=TRUE,prob=c(.5,.5))
hist(cisr,col='#D18532',xlab='Max CisR (ug/mL)',ylab='Number of progeny',
main='cis1 x wt progeny',border=NA,ylim=c(0,50))
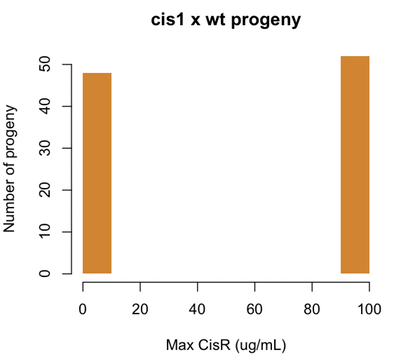
Now if we identify a CisR strain out in nature, and cross it with wild-type, we might find the following:
set.seed(1)
cisr = pmax(pmin(rnorm(n=100,m=50,s=30),rep(100,100)),rep(0,100))
hist(cisr,breaks=10,col='#DB2929',xlab='Max CisR (ug/mL)',ylab='Number of progeny',
main='CisR wild strain x wt progeny',border=NA,ylim=c(0,50))
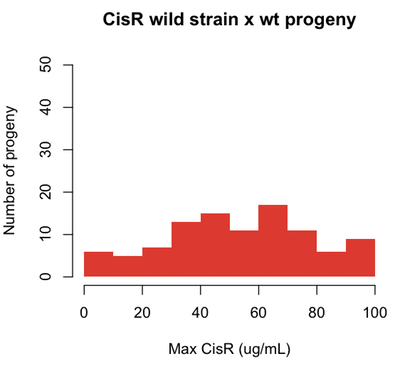
For traits that can be quantified - like the dose of cisplatin to which a cell is resistant - the genes that affect the phenotypes are called quantitative trait loci (QTLs).
A locus is the region of a genome that contributes to a phenotype.
The average human has a SNP at about 0.1% of sites in the genome. For yeast the figure is more like 0.2%. In a typical yeast cross, one can genotype >60,000 SNPs in order to identify QTLs. One way to do this is by whole-genome sequencing. As an example, consider a cross of “strain 1” - our wild strain resistant to up to 100 μg/mL of cisplatin, and “strain 2” - our laboratory strain sensitive to cisplatin. We cross strain 1 (CisR) × strain 2 (CisS), dissect tetrads, identify CisR segregants, and then perform whole-genome sequencing. Most studies will perform sequencing on ~100 progeny, some have done close to 1000. Through sequencing one can figure out which genomic regions from strain 1, the CisR strain, are transmitted the CisR progeny. The principle of this approach is diagrammed below:
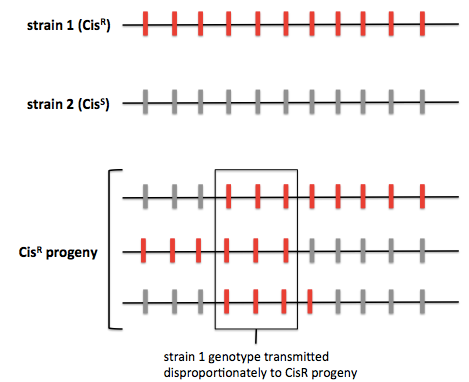
Another approach is pooled linkage analysis, also known as bulk segregant analysis. This is the same except instead of genotyping individual progeny, you pool the CisR progeny and pool the CisS progeny and count the allelic depth at each site. (For quantitative traits this presumably involves setting some threshold for inclusion in each pool, with a concomitant loss of statistical power).
One landmark study looked at two yeast strains, known as S288c and SK1 [Deutschbauer & Davis 2005]. In S288c, only 5% of diploids will sporulate, and in SK1, >95% of diploids will sporulate. When these two strains were crossed, the progeny followed a continuous distribution and clearly did not represent 2:2 segregation. They performed a cross with the following goals:
- Identify QTLs
- Identify the relevant gene at QTLs
- Identify the causal variant in each gene, dubbed the quantitative trait nucleotide (QTN)
- Quantify the contribution of each QTN
Once you have identified the QTLs, you can perform gene replacement for each variant at that site individually in order to identify the causal SNP and quantify its contribution. For instance, Deutschbauer and Davis used gene replacement to insert each SK1 variant at each QTL into the S288c strain.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.