Biolit 04: bacterial pathogenesis
These are my notes for week 04 of Harvard’s BBS 230 “Analysis of the biological literature” course (September 23-25, 2014). This includes my own reading and analysis of the papers, as well as my notes from both discussion sections.
The papers for this week are [Alto 2006] and [Huang & Sutton 2009].
Context
Bacterial effector proteins
As explained in week 01, type III secretion systems (T3SS) are a feature of certain gram-negative bacteria that evolved from flagella and retain structural similarity to flagella. They are basically a pore-forming syringe that can inject effector proteins from the bacterial cytoplasm (inside the inner membrane) into the host cytoplasm. Sometimes the purpose is to let the bacterium gain entry into host cytosol - e.g. for Shigella. Other bacteria use them to create vacuoles in the host cell - e.g. Salmonella’s SifA. Though we understand how some effector proteins work, many remain enigmatic.
For any given effector protein, a question will be, does it replace, mimic or hijack a host pathway? And how does it do that, at the molecular level. These papers were both efforts to answer those questions for one class of proteins which share a WxxxE motif, represented by three members: Shigella’s IpgB1 and IpgB2, and enterohemhorragic E. coli’s Map.
Actin
Neutrophils chase invading bacteria via chemotaxis - by extending lamellipodia, which are created by actin polymerization creating protrusions which push the cell forward. They use these to engulf and destroy bacteria.
If bacteria are successful in invading the host cell, they can polymerize host cell actin to push themselves along.
They do this by making their tail edge mimic the leading edge of a lamellipodium. They do this by mimicking a protein called Wasp (encoded by the human gene WAS) which activates Arp2/3 (ARPC2 and APRC3).
Lamellipodia are blunt protrusions thanks to Arp2/3 branching and filamin cross-linking, where as filopodia form finer finger-like protrusions as shown here:
There are yet other types of actin fibers, such as stress fibers.
GTPases
Recall that GTPases in general are facilitated by guanine exchange factors (GEFs) which replace GDP with GTP in the GTPase’s grasp - placing the GTPase in the “on” position - and GTPase activating proteins (GAPs) which encourage the GTPase to hydrolyze its GTP to GDP, placing it back in the “off” position. This is diagrammed below, using Rac GTPases (a sub-family of Rho GTPases) as an example:
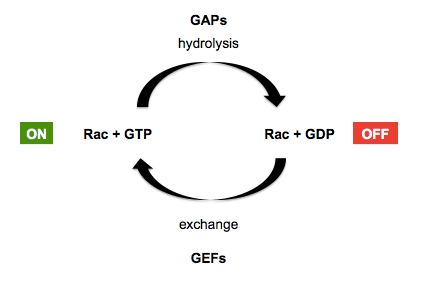
In the early 1990s, several proteins including rac, rho and Cdc42 were purified and injected into cells, demonstrating their ability to induce actin fiber formation [e.g. Ridley & Hall 1992, Ridley 1992, Nobes & Hall 1995]. RhoA mutations and fusions are now known to be common in gastric cancers
The story for this week
By 2006, certain other bacterial effector proteins were reported to act as GEFs to activate host GTPases — a quick Google Scholar search revealed Salmonella typhimurium’s SopE2 [Stender 2000] and Burkholderia pseudomallei’s BopE [Stevens 2003] to name two examples. Therefore, when PI Jack Dixon and his postdoc Neal Alto set out to identify the mechanism of the WxxxE bacterial effector proteins Map, IpgB1 and IpgB2, a reasonable prior expectation was that they would prove to be GEFs too. Instead, in [Alto 2006] they concluded that these three proteins actually acted as GTPase mimetics themselves, despite lacking any homology or obvious structural similarity to GTPases. The fact that this conclusion was so surprising is likely what got [Alto 2006] into Cell.
However, the conclusion was incorrect. Later that same year, two different groups reported that Legionella pneumophila’s DrrA (also known as SidM), another WxxxE protein (it contains the sequence WQEWE at positions 62-66), was a GEF [Machner & Isberg 2006, Murata 2006]. The truth was that Map, IpgB1 and IpgB2 actually all acted as GEFs as well, and after becoming a PI himself, Neal Alto set out to set the record straight with a crystal structure of Map bound to its host target, the GTPase Cdc42 [Huang & Sutton 2009]. In setting out to disprove his earlier work from the Dixon lab, Alto was following in the footsteps of Kim Orth, another former Dixon postdoc - see week 01.
Alto 2006: Identification of a bacterial type III effector family with G protein mimicry functions.
Big picture
This paper claims that:
- Shigella’s IpgB2 mimics RhoA, inducing actin stress fibers
- Shigella’s IpgB1 mimics Rac1, inducing lamellopodia
- Enterohemhorragic E. coli’s Map mimics Cdc42, inducing filopodia
Figure by figure
The paper begins with a BLAST search for proteins homologous to Map. They found 24 sequences from 11 distinct proteins (Figure 1A). They found that only two residues were perfectly conserved, in a WxxxE motif, but there was also a structural similarity, with 6-8 alpha helices in each protein. To confirm that the WxxxE motif was important, they transfected HEK293A cells with IpgB2, and saw stress fibers similar to what had been reported in the literature for RhoA activation. These stress fibers did not appear if the W or E residues were mutated. Two other random mutations did not prevent stress fiber induction. The stress fibers are imaged using phalloidin, an Amanitas death cap toxin that binds actin with great specificity.
Next they asked whether IpgB2 was epistatic to RhoA, meaning whether it was in the same pathway. They transfected HEK239A cells to express botulinum toxin to inhibit RhoA, B, and C, and then expressed IpgB2 as well. IpgB2 appeared capable of inducing stress fibers even when the Rhos were inhibited (though in a very close look at Fig 2B it appears the intensity of the stress fibers is reduced somewhat when RhoA is inhibited). They took this to suggest that IpgB2 was independent of RhoA, and furthermore that it was sufficiently structurall different so as to not be inhibited by botulinum toxin itself. Some bacterial effector proteins are actually evolved from host proteins for which bacteria managed to steal a copy of the host gene; the fact that IpgB2 bore no sequence similarity to RhoA and also lacked enough structural similarity to be inhibited by botulinum toxin suggested it had evolved independently.
Next they set out to do a yeast two-hybrid screen. A typical two-hybrid screen involves fusing a protein of interest (“bait”) to the DNA-binding domain of GAL4, and every member of a cDNA library of interest (“prey”) to the transcription-activating domain of GAL4. If and only if a probe binds the bait, the two pieces of GAL4 will come into close enough proximity to activate transcription of a reporter gene under control of a Gal4-dependent promoter. By making the reporter gene something essential for life under certain media conditions - say, a histidine biosynthesis gene when yeast are grown on histidine-deficient media - you can select for cells that express a cDNA whose protein product does bind the bait. Then you sequence the cDNA in the surviving cells to see which cDNAs they were. Once you have hits, you can confirm by isolating clones and re-validating in yeast, then moving on to co-immunoprecipitation and other biochemical methods.
In this study, they found that IpgB2 expression in yeast was lethal, which made it tough to do a two-hybrid screen with IpgB2 as bait. So they mutated the E residue from the WxxxE motif, hoping this would render IpgB2 catalytically inactive but still able to bind its binding partners. In their screen they identified CRIK, a kinase which Rho activates (Fig 3B). CRIK has homology to ROCK proteins, so next they performed co-immunoprecipitation to confirm their hypothesis that IpgB2 would bind ROCK as well. They expressed IpgB2 fused to GFP in HEK239A cells, and then immunoprecipitated by mixing cell lysates with anti-GFP antibody conjugated to beads, and washing away anything unbound to the beads. Their blot in Fig 3C shows that ROCKs were immunoprecipitated by this method. In Figure 3D they measure ROCK kinase activity of their various immunoprecipitates, and show that IpgB2, co-immunoprecipitates with ROCK kinase activity. Map also has the WxxxE motif, yet doesn’t pull down ROCK activity, so the WxxxE motif must not be what binds ROCK. This activity is inhibited by the ROCK inhibitor Y27632, so it is definitely ROCK. Moreover, inhibiting ROCK prevents IpgB2 from inducing actin stress fibers (Fig 3E) which indicates that ROCK lies downstream of IpgB2 in the pathway by which IpgB2 induces actin stress fibers. Next they pulled down FLAG-tagged mDia1 (DIAPH1 gene in humans), another Rho substrate which has formin homology domains (FH1 and FH2) which are involved in promoting actin polymerization, and showed that this pulled down IpgB2 (Fig 3G). mDia1 ΔN3 did not do this, suggesting the N3 domain is required for binding.
It is somewhat mysterious that CRIK does not appear anywhere in Figure 3. After they spent months on the yeast two-hybrid screen to identify CRIK, they immediately depart from CRIK in favor of two related candidate proteins, and never really explain why. The existence of ROCK inhibitors may be one factor; it is possible they tried to co-immunoprecipitate CRIK and were unable to do so and did not report it.
They expressed a variety of mutants of IpgB2 in yeast to see which were toxic. Of the few options they tried, only mutating the WxxxE motif or introducing a “ΔN84” mutation (which they never explain but was probably a deletion of 84 N-terminal residues) restored yeast viability. They screened yeast deletion mutants to try to find genes whose deletion restored viability in cells expressing IpgB2. In an effort to confirm their hits, they claimed a correlation between gene signatures detected by microarray (Fig 4E).
Then they examined the end consequences of IpgB2 and Map, which is that they induce lamellipodia and filopodia (Fig 5). Map transfection results in phosphorylation of JNK and c-Jun in HEK239A cells, but evidently does not phosphorylate ERK (Fig 5I), although since there is no positive control for ERK1/2 phosphorylation it is impossible to rule out ERK activation.
Next they sought to identify binding partners of a C-terminal sequence conserved across many WxxxE proteins, which is VQDTRL in Map. They found Ebp50, and confirmed by radiolabeling Ebp50 (Fig 6E-F) and by co-immunoprecipitation (Fig 6G) that it does bind Map. Ebp50 is also known to bind the VQDTRL sequence in CFTR, which is a cell surface membrane protein. TMutating this sequence abolished the filopodia-inducing activity of Map (Fig 6H-I). Together these results suggest that Map’s subcellular localization is important for its activity.
Finally, in Figure 7 they move to in vivo experiments with live bacteria - everything until now was just using transfections. They show enteropathic E. coli on HeLa cells (Fig 7A-D) though E. coli are extracellular, so they are not in the cells. Mutant versions of Map do not induce filopodia, and knocking down Ebp50 reduces filopodia formation.
Limitations
Some specific limitations are:
- To show that Map is independent of Cdc42, it would have been nice to show that Map can induce filopodia without Cdc42. Unfortunately Cdc42 knockout is cytotoxic. However, they could have used a hypomorphic allele. If Map is truly independent of Cdc42, it should function the same in the presence of hypomorphic Cdc42.
- They never directly tested possible GEF activity of IpgB2 or Map.
- Figure 2 could have included a mammalian GEF as an additional control. Presumably the reason why IpgB2 still induced stress fibers when the Rho proteins were inhibited was that the inhibition was not absolute, and even a small amount of free Rho protein, if activated by IpgB2, could have a powerful effect. If so, then overexpressing a mammalian GEF might have had the same effect as IpgB2.
- In Figure 3 they could have blotted for Rho as well as for CRIK and ROCK. They would presumably have seen that Rho was also co-immunoprecipitated, suggesting that it is the intermediary which binds both IpgB2 and CRIK/ROCK.
- They never tried to reconstitute anything in a cell-free reaction - for example, they never directly measured the ability of purified IpgB2 to hydrolyze GTP.
Overall, the paper also surveys a vast expanse of topics - three different bacterial proteins and their host targets - without exhaustively tackling any one of them. The text does little to transition between different experiments or provide a unifying logical flow.
Huang & Sutton 2009: Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics
As mentioned above, shortly after [Alto 2006] came out, two other groups showed that DrrA, a different WxxxE protein, was a GEF. In 2008, Alto and Dixon themselves were involved in an effort to crystallize Salmonella SifA, another WxxxE protein, which revealed that it too must be a GEF [Ohlson 2008]. Based on this, Alto hypothesized that Map was actually a GEF that bound Cdc42.
Figure by figure
To purify the two proteins, they expressed MBP-tagged Map and GST-tagged Cdc42. They confirmed that the two proteins bound one another, and that the WxxxE motif was critical for binding (Fig 1A). They then tested the ability of Cdc42 to bind GTP, and found that the rate of binding was enhanced by the presence of Map (Fig 1B). This was direct biochemical evidence that Map was a GEF. Therefore they set out to crystallize the complex, and structures are shown in Fig 1C-D.
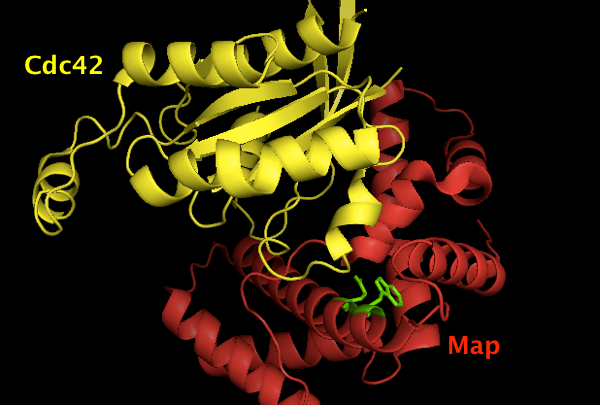
The structure is deposited as [PDB# 3GCG]. Using PyMOL I created this image of Map (red) binding Cdc42 (yellow), with the Map W74 and E78 residues shown in green. Apparently they are important for the binding interface, though it is not visually obvious why it should be so, and the paper simply says they “contribute many interactions between them (not shown)”.
fetch 3gcg
hide everything
show cartoon
color red, chain B # Map
color yellow, chain A # Cdc42
show sticks, chain B and (resi 74 or resi 78)
color green, chain B and (resi 74 or resi 78)
Next they used a panel of Map mutants to confirm that a a catalytic loop was crucial for GEF activity (Figure 2A), and that other regions were crucial for binding to Cdc42 (Figure 2B).
They compare their structure to other reported structures of two other GEFs bound to Cdc42. The interaction of Map and Cdc42 looks very similar to that of bacterial effector SopE and Cdc42 (Figure 3B). In contrast, the endogenous human GEF ITSN binds to Cdc42 with a totally different protein-protein interaction interface, though the key contacts at the “switch” sites are still the same, indicating that Map and SopE are non-homologous mimics of ITSN.
Map and SopE are both specific for Cdc42 and not other GTPases, which raises the question of how specificity is achieved. Figure 4 zooms in on a “beta patch” interface that may help to confer this specificity, and shows how RhoA and Rac1 would be expected not to fit here. Mutating the specificity-conferring “beta patch” on Cdc42’s side abolishes the interaction (Figure 5C), while swapping Cdc42’s beta patch into Rac1 causes Map to bind Rac1. This confirms that the “beta patch” is necessary and sufficient for specificity. Moreover, if you mutate the “beta patch” of Cdc42 or Rac1 to be that of RhoA (IpgB2’s target), you can get IpgB2 to exert GEF activity on Cdc42 or Rac1 (Figure 6C).
Discussion
The [Alto 2006] paper reads well, and in fact does contain many true observations. At first glance nothing seems obviously wrong with it. How, then, can one avoid reaching incorrect conclusions, or believing others’ incorrect conclusions?
As discussed above, there are a number of controls and experiments that [Alto 2006] could have added at the micro level which would have helped. At the more macro level, one could describe most of the experiments in [Alto 2006] as being confirmatory - they aimed to confirm the hypothesis that IpgB2 et al were GTPase mimetics. None were specifically designed to falsify the hypothesis. So one moral of the story is to always be suspicious of your own hypothesis and try as hard as you can to disprove it. Alternatively, you could argue that all of the data presented in [Alto 2006] is in fact both accurate and consistent with the true story from [Huang & Sutton 2009], and that it is simply the interpretation which is incorrect, so this argues for being more conservative in interpreting one’s data.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.