Molecular Biology 13: 'Homologous recombination and non-homologous end-joining'
These are my notes from lecture 13 in Harvard’s BCMP 200: Molecular Biology course, delivered by Johannes Walter on October 6, 2014.
In lectures 11 and 12 we covered DNA damage repair mechanisms which use the complementary strand as the source of information on the correct sequence. In this lecture we’ll cover homologous recombination (HR), which uses the sister chromosome as a source of information, and non-homologous end-joining (NHEJ), which has no source of information. These two types of repair address double-strand breaks (DSBs). DSBs are dangerous because repairing via NHEJ can lead to translocations between two chromosomes, or loss of a chromosomal arm, either of which can lead to cancer.
Double-strand breaks
Sometimes the cell intentionall makes DSBs. During meiosis an enzyme called Spo11 causes DSBs in order to promote recombination and thus create a source of diversity. However, most DSBs are unwanted and result from exogenous sources such as ionizing radiation, oxidative free radicals, mechanical stress, or Topo II failing to rejoin the duplexes it cuts. All of the above are called “two-ended DSBs”. In contrast, a “one-ended DSB” results frmo a nick in the leading strand template during replication. This is “one-ended” because there isn’t a second end, so there is no opportunity for non-homologous end-joining. Such nicks can result from endogenous sources of damage or from excision repair mechanisms as discussed in the last two lectures.
Non-homologous end-joining (NHEJ)
The NHEJ pathway is reviewed in [Lieber 2010]. The first protein to respond to double-strand breaks is Ku70/80, which is present at very high concentrations in cells and thus is recruited very quickly to DSBs. It in turn recruits DNA-PKcs (kinases) and then Artemis (which removes damaged ends) and end-processing enzymes. DNA pol λ and μ will then re-extend the ends. DNA pol λ can actually extend DNA without a template in order to give you sticky ends you can anneal. Ligase IV is a versatile ligase which can ligate pretty much any DNA sequence, and it will ligate as soon as the strands are annealed. Artemis will then trim any hanging flaps.
This process is extraordinarily error-prone - it may add or delete genetic information and can cause chromosomal translocations. However, it is believed to be preferable to no repair in many circumstances.
Homologous recombination (HR)
We’ll introduce HR in the context of its ability to fix two-ended DSBs. At both of the double-stranded ends facing each other, the 5’ strand is resected (trimmed back). A recombinase (RecA in bacteria, Rad51 in eukaryotes) will then bind the single strand. This complex is called a presynaptic filament. The recombinase will then perform strand invasion whereby it base-pairs the single strand with the complementary strand of the homologous duplex. This forms a “D-loop”. The single strand is then extended, using the complementary strand as template, until it reaches a region homologous to the 3’ end of the opposite side of the break. Then the second end can be recaptured, after which gap filling and ligation occur (not pictured).
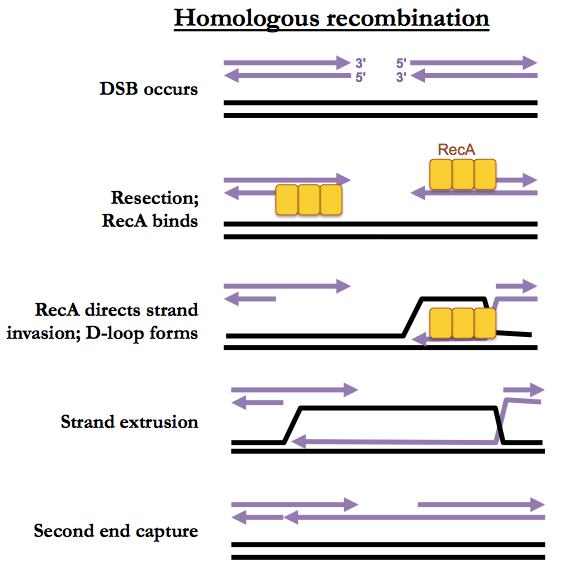
The above pathway is called synthesis-dependent strand annealing. A more complicated pattern of HR involves Holliday junctions but that pathway is preferred only during meiosis and is otherwise rare.
Note that in G1 phase, a double-strand break could only be repaired by HR if it were to utilize the other parent’s chromosome as a template. However during G1, sister chromosomes are not physically adjacent, so this is difficult. Instead, NHEJ is preferred during G1. During S phase, by contrast, identical sister chromosomes are adjacent to each other, held together by cohesin.
A biochemical assay for RecA-dependent strand exchange involves introducing a pre-formed pre-synaptic filament along with a linear piece of dsDNA, and monitoring for strand exchange resulting in a nicked circular dsDNA and a linear single strand. These can be distinguished by their electrophoretic mobility. RecA is a AAA+ ATPase which can form a helix around either ssDNA or dsDNA, with each RecA molecule interacting with about 3 nucleotides, and about 6 RecA molecules per DNA helical turn. A seminal experiment involved crystallizing RecA bound to ssDNA [Chen 2008]. This required fusing 6 copies of RecA end-to-end, and mutating the binding site at the N-most RecA, so that the protein being crystallized was a single larger protein rather than a heterogeneous filamentous collection of monomers. In spite of all these mechanisms, this 6-mer was still catalytically active.
In this biochemical assay, Rad51 is far less active than RecA.
BRCA2 example
Mutations in BRCA2 lead to a ~80% lifetime risk of breast cancer. Its mechanism was discovered through the convergence of many different experimental approaches. First, a yeast two-hybrid screen revelead that Brca2 interacted with Rad51 [Sharan 1997]. BRCA2-/- cells are hypersensitive to radiation-induced death [Abbott 1998]. Cell biology experiments showed that upon irradiation of wild-type cells, Rad51 forms punctate foci that colocalize with markers of DSBs, while this reaction is completely suppressed in the absence of Brca2 [Yuan 1999].
The above assays are not specific to HR and it was difficult to exclude a role in NHEJ. An assay more specific to HR involves a cassette carrying two copies of GFP, both of which are inactviated by different mutations [Pierce 1999]. Digestion with the I-SceI restriction enzyme results in a double-strand break which creates an opportunity to create a functional copy of GFP via HR. In this assay, there is far less HR activity in the absence of Brca2 [Moynahan 2001].
Brca2 is 3500 amino acids - too large to crystallize - but a few domains have been crystallized. It contains 8 BRC repeats which bind Rad51 [Pellegrini 2002] and has both ssDNA- and dsDNA-binding domains [Yang 2002]. Full-length Brca2 is highly insoluble but was finally purified in 2010 by fusion with other, better-behaved proteins [Jensen 2010, Liu 2010, Thorslund 2010]. This purification allowed biochemical reconstitution of Brca2’s activity and determination of the stoichiometry between Brca2 and Rad51. RPA, another ssDNA-binding protein,, can out-compete Rad51 alone for binding to ssDNA but cannot out-compete Rad51 plus Brca2 [Jensen 2010]. This suggests a model in which RPA opportunistically binds to ssDNA following the resection step of HR, and that Brca2 is then required to recruit many copies of Rad51 and allow them to overcome RPA to bind to the ssDNA.
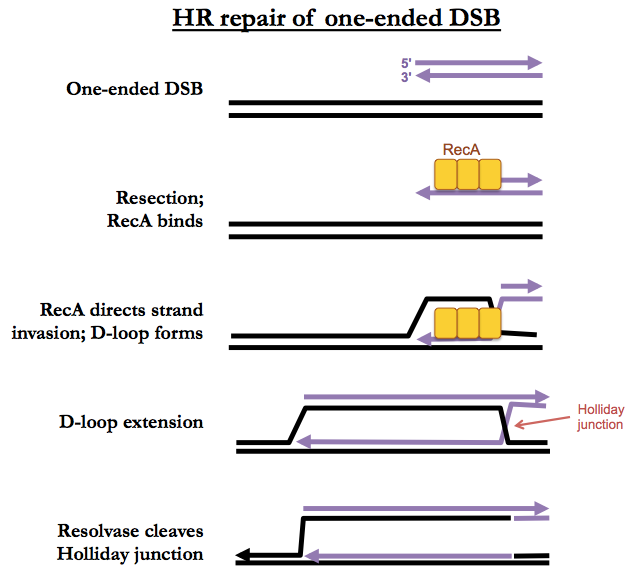
One-ended DSBs
The beginning of one-ended DSB repair looks just like two-ended DSB repair, with strand invasion, D-loop formation and D-loop extension. However there is no second end to capture, so D-loop extension just proceeds, forever, creating both a leading and lagging strand, leaving a Holliday junction behind it. Eventually an enzyme called resolvase will simply cleave the Holliday junction, leaving you with just a single replication fork. This may or may not result in a crossover.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.