Genetics 17: 'Forward genetic screens in Drosophila'
These are my notes from lecture 17 of Harvard’s Genetics 201 course, delivered by Mitzi Kuroda on October 29, 2014.
Introduction
Forward genetics means using mutagensis to create random mutations, then searching for the genotypes that underlie the resulting phentoypes. (The opposite, reverse genetics, means creating targeted mutations in known genes order to see what the phenotype might be, for instance in knockout mice). This lecture will cover forward genetics in Drosophila.
Facts about forward genetic screens in Drosophila
Mutant screens revealed mutants that are missing all of the odd-numbered body segments, and other mutants missing all the even-numbered body segments. This led to the conclusion that:
the process of segmentation involves at least three levels of spatial organization: the entire egg as developmental unit, a repeat unit with the length of two segments, and the individual segment
In Drosophila, transgenics are created by microinjection into the posterior end of the embryo, where the germ cells live.
It was predicted that loss-of-function mutations in genes specifying the Drosophila body plan would have embryonic lethal phenotypes (“failure to develop”). The ambitious goal was to find all genes involved in the body plan, which would require a large number of flies. Challenges included:
- How to homozygose the mutations?
- How to create a stable stock carrying the mutation
A typical cardboard box used in fly labs holds 100 vials of flies, and the scale of screen here requires 10,000 lines of flies, so 100 boxes. So this is quite an organizational effort even in Drosophila. This ambitious goal would be difficult to achieve in mnay other model organisms. Mice would be prohibitively space- and time-consuming. In C. elegans you could operate at the scale necessary to get the number of organisms and mutations that you need, but C. elegans don’t have such a dizzying array of wonderful phenotypes as Drosophila do. Zebrafish have awesome phentoypes, and maybe you could do this with them.
In this screen, as they looked at more and more lines of flies, the number of body plan mutants increased linearly, topping out at 272. The number of loci identified plateaued at 48, so that by the end they were looking at multiple mutants in the same loci.
Note about nomenclature: in Drosophila sometimes the P0 generation is called G0.
In Drosophila, once females have mated once they will be less interested in mating again, and will carry sperm and/or fertilized embryos around with them. Therefore, for a controlled cross you need to use virgin females, which are denoted with the Mercury symbol (☿). Newly enclosed females are guaranteed virgins for about 8 hours at 25°C. At this stage you can segregate them into their own male-free vial.
How to screen for embryonic lethal mutants in Drosophila
Today we will discuss the design of an F3 screen for developmental genes on chromosome 2. Because screening for lethal mutations requires balancer chromosomes (see lecture 16), you can only look at one chromosome at a time. In principle you could use flies which have balancer chromosomes for each of their chromosomes, but in practice such flies are quite sick and difficult to work with.
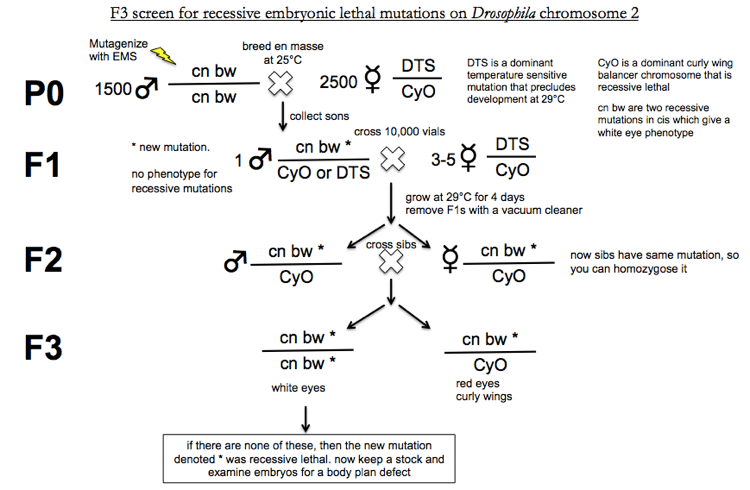
A few comments on the above graphic.
First, the reason why all F2s have the genotype cn bw * / CyO is as follows. When you cross the F1s the Punnett square looks like this. Sperm haplotypes are across the top, egg haplotypes are across the left.
| cn bw * | CyO | DTS | |
|---|---|---|---|
| DTS | |||
| CyO | cn bw * / CyO |
You’re growing at 29°C for 4 days, so any offspring with at least one DTS allele are dead. In addition, CyO is recessive lethal, so CyO/CyO are dead too. Only the cn bw * / CyO progeny are viable.
Similarly, in the F3 generation, all CyO/CyO progeny will be dead.
This screen resulted in 4217 lethal mutants, of which 1620 were embryonic lethal. The researchers developed cuticle prep as a quick way to look at the body plan. Careful phenotypic analysis revelead three levels of spatial resolution (per the quote above). They found gap, pair-rule and segment polarity mutants. 272 body plan mutants were identified. 2722 = 73,984, prohibitively many for exhaustive complementation tests. Therefore they only conducted complementations tests on mutants that had similar phenotypes. Based on this they found 48 loci with 2 or more alleles, and an average of 5.4 alleles per locus. Dr. Kuroda contacted Dr. Weischaus to ask whether any additional body plan mutants had been identified since the original study, and the answer was no - so this screen apparently identified all recessive lethal body plan mutants.
From mutants to genes
Ideally, you want to start with isogenic inbred lines. Drosophila are more polymorphic than other model organisms and it is hard to get perfectly inbred lines that are 100% homozygous, but you try to get them as homozygous as possible, at least for the chromosome of interest. To maximize homozygosity, you want to grow your 1500 males for the P0 stage of the screen from as recent a most common ancestor as possible.
You can then perform sequencing to identify genetic variants that differ from the parental line.
Transgenic complementation
The gold standard for proving that a mutation is causal is transgenic complementation, borrowing some reagents from bacteriophage genetics. Prepare a plasmid with a wild-type copy of the candidate gene, plus eye-GFP (that’s GFP under an eye-specific promoter), and an attB site. Inject the plasmid into the posterior of embryos that express ØC31 integrase and have an attP site in their genome. (Flies with this genotype are available commercially. They were originally created using random transposon insertion of attP sites and then subsequent mapping of the inserted attP sites). Integrase will integrate the plasmid into the genome at the attP site. The injected fly will not have GFP glowing in its eyeballs because the plasmid is only in its posterior. Its transgenic progeny, however, will have GFP in their eyes. Such progeny are assumed to also have received the wild-type gene. Now cross them into your recessive lethal mutant stock to test for complementation. If you originally were getting no white-eyed flies in your F3s (see above graphic), and now you DO get white-eyed flies, then the wild-type gene successfully complemented the mutation.
A consideration for transgenic complementation is whether to use a full genomic copy of the gene, or a cDNA? The safest option would be to use a genomic copy because it is more likely that all the regulatory elements are in place. However, sometimes the size of the full genomic locus is prohibitive. In addition, cDNAs have some advantages, notably that you can aim for expression specifically in your tissue of interest (using GAL4-UAS??).
In coming years, it is to be expected that more Drosophila work will use CRISPR/Cas9 and homologous recombination to perform gene replacement into the endgenous locus rather than using transgenes. In addition, “flies will become more like yeast” (not sure what this means).
Other examples of mutants discovered in various types of screens
Circadian rhythm screens identified period (per), a transcription factor. Mutations in the human ortholog PER2 cause familial advanced sleep phase syndrome [OMIM #604348].
Learning and memory screens. Wild-type flies will avoid an odor associated with a prior bad experience. Mutants deficient in this behavior turn out to have mutations in enzymes affecting cAMP signaling in neurons.
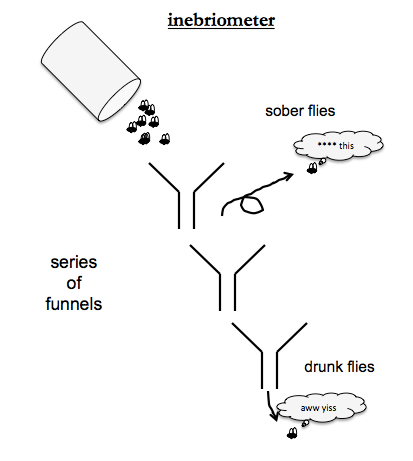
Ethanol tolerance screens used an “inebriometer” - an apparatus of a series of funnels, where each funnel spits out its contents onto the rim of the next one. Sober flies will get tired of sliding down the funnels and just fly away, but drunk flies, like drunk humans, love sliding down slides. So at the bottom of all the funnels you get the drunkest flies. By mutagenizing flies and the exposing them to ethanol vapor, people identified mutants with low ethanol tolerance. The causal gene was named CHEAPDATE and encodes a neuropeptide involved in regulating cAMP.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.