Protein folding 04: Formation of alpha helices
These are my notes from week 4 of MIT course 7.88j: Protein Folding and Human Disease, held by Dr. Jonathan King on February 26, 2015.
Assignment 3
The readings for this week are chapter 15 of Branden & Tooze on immunoglobulins, and [Richardson & Richardson 1988, Marqusee & Baldwin 1990, Harbury 1993].
Richardson & Richardson 1988
At the time, figuring out where you define the start and stop of an alpha helix was a major problem. They considered two possible models for defining where a helix starts and stops. A model based on φ and ψ angles was rejected. Instead, they chose a model where the last amino acid whose alpha carbon (αC) falls within the cylinder defined by the helix, is considered to be the N-cap or C-cap, meaning the first or last residue in the helix. This model was preferred because it gave stronger signals of preference for which amino acid should be the cap. Proline is the amino acid most rarely seen in alpha helices, for two reasons: 1) it cannot rotate around its N-C bond, and 2) its N is not protonated, so it cannot participate in the hydrogen bonding that defines the alpha helix backbone. D and E, the negatively charged / acidic residues, are preferred near the N terminus; K and R, the positively charged / basic residues are preferred near the C terminus. This is consistent with the dipole moment model of alpha helix formation.
A related and interesting set of analyses were done by [Presta & Rose 1988]. They took alpha helical structures and modeled different amino acid substitutions at the N and C termini in silico. They found that it was necessary that near the ends there be side chains which can satisfy some of the hydrogen bonding requirements of the backbone, and then another residue which can shield those side chains.
Marqusee & Baldwin 1990
This book chapter chronicles some of the landmark experiments and observations which informed our understanding of how sequence drives alpha helix formation. This chapter was written in 1990, when hopes were high that de novo design of peptides was just around the corner. They review the experiments of Baldwin’s group with RNAse A C-peptide, which are covered in more detail in the lecture notes below. They also discuss a de novo designed peptide consisting of all alanines except for repeating units of E and K organized along i±4n or i±3n ridges. Helix formation was optimal with E and K in the i±4n ridge, and this suggested that the formation of salt bridges, and the dipole moment of the protein, were important.
Their designed peptide was very different from any peptide seen in living organisms. Helices are almost never seen naked and do not have charged residues on both sides of the helix - instead, they have one hydrophobic face.
Harbury 1993
The authors created a series of mutants of the GCN4 coiled coil, changing the residues in the a and d positions either to all V, all I, all L, or various pairwise combinations. When a was all I and d was all L, you got a dimer. but when a was all L and d was I, you got a tetramer. When both were I, you got a trimer. When V was involved, you got a mixture of species. These dramatic differences showed that specific residues - and not just the hydrophobicity of the interaction surface - mattered a lot.
Sadly, 22 years later, this work remains “cutting edge” in the sense that we still don’t understand any better why each residue has such different effects on multimerization. One limitation of the paper is that with just 2D images and words, it is very difficult to convey exactly what drives the differences in multimerization. You really need space-filling models to be able to visualize that.
Sequence determinants of alpha helical folding and unfolding
Pauling had the early insight that when the O (δ-) and N (δ+) of a peptide backbone hydrogen bond with one another, that removes the polar aspects of the backbone from further interactions, thus making the peptide behave in a largely hyrdophobic manner. The backbone atoms thereby participate in Van der Waals interactions, excluding bulk water.
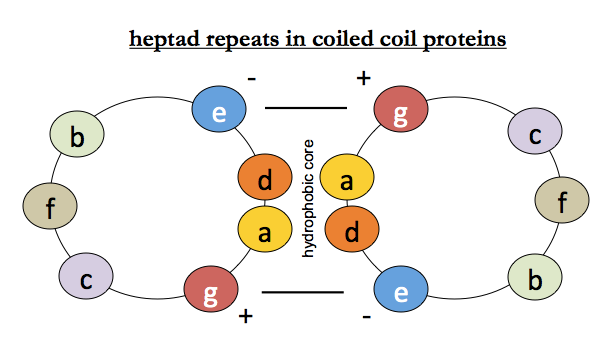
For coiled coils, we additionally know that there is hydrophobic packing between helices (positions a and d) and that salt bridges surrounding them (positions e and g) stabilize the interaction - recall this diagram:
Equilibrium unfolding and refolding of tropomyosin: the local alignment hypothesis
In tropomyosin from rabbit heart muscle, coiled coil pairs form rods about 40 nm long, with each helix consisting of 284 residues (~40 heptad repeats). No one has ever crystallized tropomyosin, but it does form fibers, so we have fairly high-resolution structural information from X-ray fiber diffraction. The coiled coil of tropomyosin purified from muscle is a heterodimer of α and β chains which differ in about 39 amino acids.
Tropomyosin can be studied as follows. The disulfide bonds can be reduced, and the dimer will retain its native conformation. You can then use circular dichroism at 222 nm (the deepest trough characteristic of alpha helices) to measure its helical content. Then use old-fashioned thermal denaturation: heat the protein and measure at what temperature it loses helical content. Specifically, if you want to do equilibrium unfolding, you stop at each unit of temperature (say, each 1° or 0.1°) and wait until the CD signal stops changing, then proceed. Once you reach total denaturation, you can do the same process in reverse to monitor equilibrium refolding.
Consider that egg white is composed predominantly of lysozyme and albumen, both of which are medium-sized globular proteins. When heated, they aggregate and become opaque, a change which is irreversible upon cooling. Milk is composed primarily of casein and lactalbumen, and when heated it will curdle, which is also irreversible upon cooling. Proteins in most warm-blooded animals function best in the 37-39°C range, and will denature at the just slightly higher temperatures associated with fever. Yet tropomyosin’s unfolding is reversible. These experiments were done by a couple, Holtzer & Holtzer [Holtzer 1983]. They posited that at temperatures of total unfolding, tropomyosin was a “random coil.”
Suppose that in refolding, each tropomyosin peptide first independently formed an alpha helix, and these subsequently interacted to form a coiled coil. However, the interface between the two helices is highly hydrophobic, and Modey and Baldwin reasoned that the exposure of this face to solvent would be very unstable. One possible handwaving solution was if the helices formed very transiently, and then would either associate if possible, or if not, then they would unfold very quickly. But someone Holtzer and Holtzer realized that you could use light scattering to monitor molecular weight during equilibrium unfolding and refolding, at the same time as measuring alpha helical content with CD. When they did this, they found that dissociation of dimers into monomers was almost perfectly coupled with unfolding of alpha helices into random coils. Thus, it was not the case that each helix formed independently before dimerization - the process was cooperative. However, there was a rub. If the helices form independently and then associate, then formation of helical content (measured by CD) should be independent of concentration, whereas dimerization should be dependent on concentration. If helix formation is cooperative, as Holtzer and Holtzer supposed, then dimerization and helical content should both be concentration-dependent. Yet Holtzer and Holtzer’s data actually showed that helix formation was independent of concentration. To explain this finding, they offered the following, still unproven, explanation. Naked alpha helices have 3.6 residues per turn, while coiled coils have 3.5 residues per turn. So two helices can align locally only at the expense of being disordered and unaligned elsewhere, until they have enough time to, bit by bit, twist each other into alignment. They posited that the tropomyosin coils have extremely high affinity for one another, such that association is quick, even at low concentrations, and that the rate-limiting step is not association, but cooperative re-folding. They proposed that this cooperative re-folding process, which they called “reptation”, proceeded by what you might call an “inchworm”-like mechanism, where different parts of the helix would melt and re-form incrementally until all residues were in alignment.
This idea of a protein readily forming a local alignment, but having difficulty finding global alignment, is analogous to the ability of collagen to form gelatin. When you make chicken soup, fat will float to the top. If it is skimmed off, you will get a thin viscous membrane across the top. That membrane is made of rendered collagen. Collagen is folded with in-register repeats, and once it has been heated and denatured, it cannot get back into register. Molecules of collagen associate with each other by aligning in register over a local region, but it can’t reach global alignment.
The RNAse S-peptide and C-peptide
Another seminal experiment examined the S-peptide of RNAse, which was a single alpha helix at the extreme N-terminus of RNAse, which was otherwise a largely β-sheet rich protein [Brown & Klee 1971]. They cleaved RNAse in one place to yield the S-peptide (residues 1-20) and the remainder of the protein, termed the S-protein (residues 21-124). The S-peptide contains residue H12, which is part of the active site, so RNAse only acquires enzymatic activity if the S-peptide and S-protein can associate with each other. At low temperatures (3°C), the isolated S-peptide had a CD spectrum consistent with an alpha helix. When heated to physiological temperatures (39°C), however, its CD spectrum went flat, implying it lost this helical content.
Baldwin, Kim and Shoemaker found that a subset of the S-peptide, residues 1-13 of RNAse (which they called the “C-peptide”) formed an alpha helix, but residues 14-20 were disordered [Shoemaker 1985, Shoemaker 1987]. They used solid state synthesis to create a C-peptide with sequence acetyl-AETAAAKFERAHA. They then tested its helicity across a range of pH. It was most helical at pH ~5. They noted that the C-peptide contains two negatively charged residues (E2 and E9) and three positively charged residues (K7, R10, H12). Notice that residues E9 and H12 are part of the same i±3n ridge. They hypothesized that the salt bridge between these residues helped to stabilize that ridge, driving formation of the alpha helix. This would explain why the peak is at pH=5. At very low pH, glutamate becomes protonated and loses its charge; at high pH, histidine deprotonates and loses its charge. A pH of 5 is right in the sweet spot where both proteins are charged. So to test this hypothesis, they mutated E9 to L, expecting they would lose the helical content. But surprisingly, the peptide acetyl-AETAAAKFLRAHA was actually even more prone to form alpha helices. Next they tried mutating E2 and H12. If they mutated H12, they got less of a decrease in helicity at high pH, but the helix was still sensitive to low pH. Conversely, if they mutated E2, they got less of a decrease at low pH, but the same behavior at high pH. Thus, although charges on both residues contributed to helix stability, they did so independently:
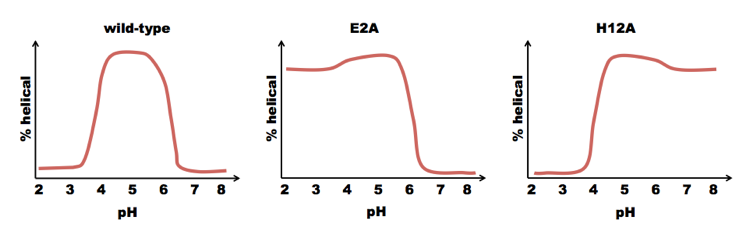
These findings supported the hypothesis that alpha helix formation dependend upon the dipole moment of the peptide. In support of this, acidic residues were mostly found at the N terminus of alpha helices, and basic residues mostly at the C terminus.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.