Organic chemistry 16: Stereochemistry - complex chirality, prochirality, topicity
These are my notes from lecture 16 of Harvard’s Chemistry 20: Organic Chemistry course, delivered by Dr. Ryan Spoering on March 9, 2015.
Prochirality and axial chirality
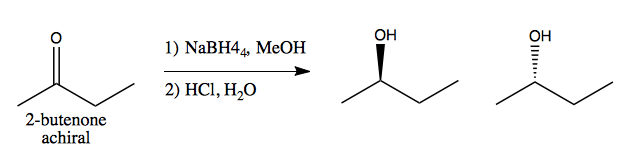
Prochirality is the idea that a set of achiral groups can give rise to chirality in a reaction. For instance, consider 2-butenone, an achiral molecule. The carbonyl “faces” are said to be enantiotopic.
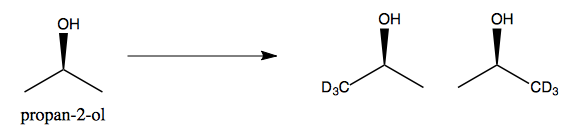
Next, consider propan-2-ol. It is achiral because its two methyl groups are equivalent. These two methyl groups are therefore enantiotopic. If we replace one - say, if we deuterate exactly one of the two methyls - then we end up with a chiral molecule.
| a face, atom or group is… | if its replacement leads to… | distinguishable? |
|---|---|---|
| homotopic | identical molecules | never |
| enantiotopic | enantiomers | in a chiral environment |
| diastereotopic | diastereomers | always |
Note that homotopic faces, atoms or groups can never be distinguished - they look exactly the same in NMR, in any reaction, etc. Enantiotopic faces, atoms or groups look the same on their own, but will react differently with chiral molecules. Diastereotopic faces, atoms or groups always appear different.
Homotopic example
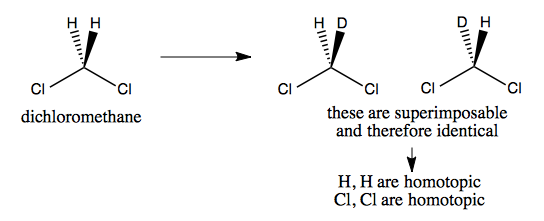
Dichloromethane’s two Hs and two Cls are homotopic, because if we deuterate one hydrogen, we still don’t get a chiral molecule.
Enantiotopic example
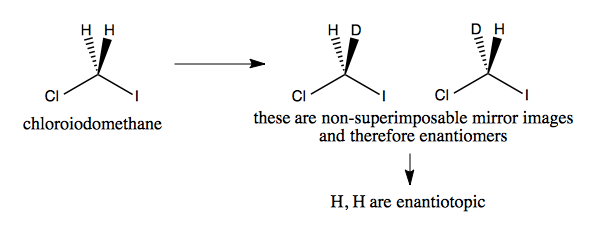
In contrast to above, if we consider chloroiodomethane, replacing one hydrogen with deuterium does create enantiomers, which could be recognized in a chiral environment (would react differentially with a chiral molecule). Therefore the H and H in chloroiodomethane are enantiotopic.
Diastereotopic example
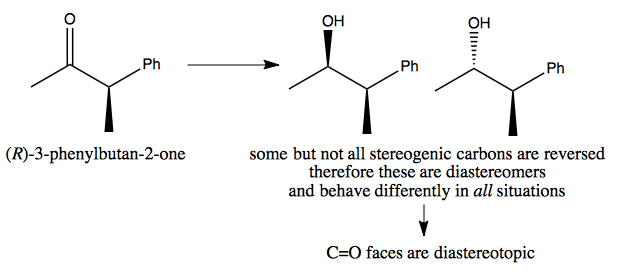
Here, the two resulting molecules are completely distinct. Therefore, reactions leading to them may have different energy profiles, and so it may be possible to design a reaction to produce only one or the other.
Here is another example where one reactant is more clearly favored over the other:
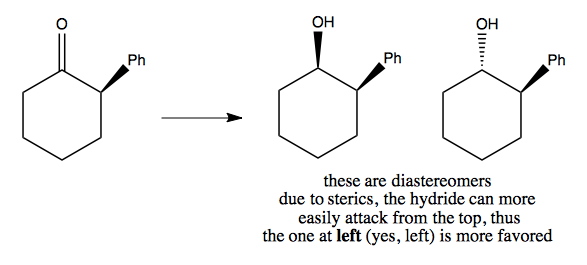
More examples
Distinguishing these three cases is not always trivial. Think through this example:
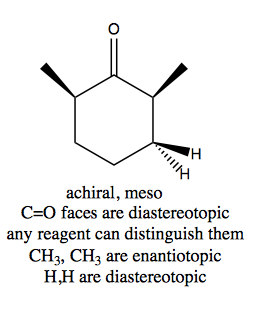
And contrast with this example:
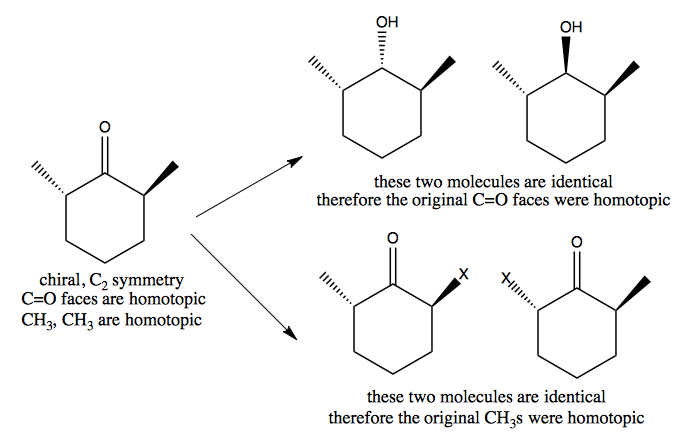
Chirality without stereogenic carbon
Heteroatoms
As introduced last time, amines can be stereogenic, but they undergo rapid Walden inversion, thus interconverting readily between enantiomers.
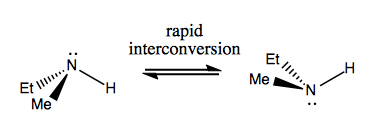
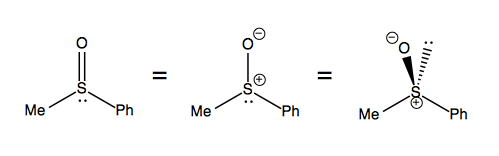
Sulfoxides, in contrast, legitimately stay as one enantiomer. This molecule is best thought of as a tetrahedral molecule with two formal charges, as depicted at far right:
Chiral conformations
As introduced last time, Gauche butane has enantiomers, but they readily interconvert:

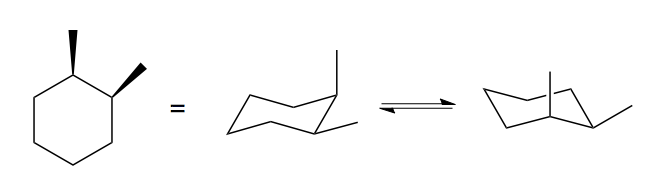
Then there’s this thing. The drawing at left is actually ambiguous, because it doesn’t tell you which chair conformation the cyclohexane is in. In fact, the two are enantiomers, but again, they interconvert readily.
Then there’s allenes, which are compounds with two adjacent double bonds at the center. Surprisingly, the bonding in allene is not planar:
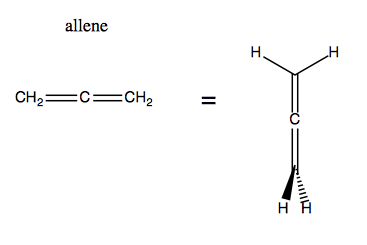
So if we start substituting, we can quickly get to a chiral molecule:
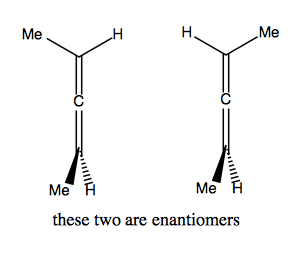
Atropisomerism
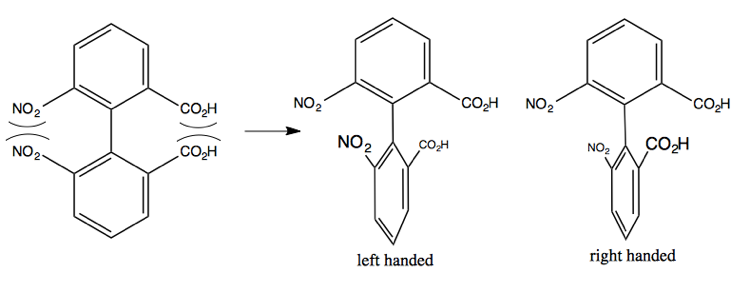
The Linderod reaction can create this molecule at left, but the steric clash between the indicated groups is so strong that it twists the central bond, breaking the conjugation between the two phenyl groups. It can twist right or left, creating two different enantiomers. Once it does so, it is energetically impossible to go back the other way, so these never interconvert. Thus, these two resulting molecules are called enantiomeric atropisomers.
Perhaps the most famous example of this is (s)-(-)-BINAP.
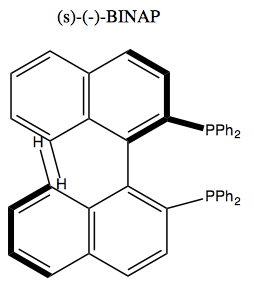
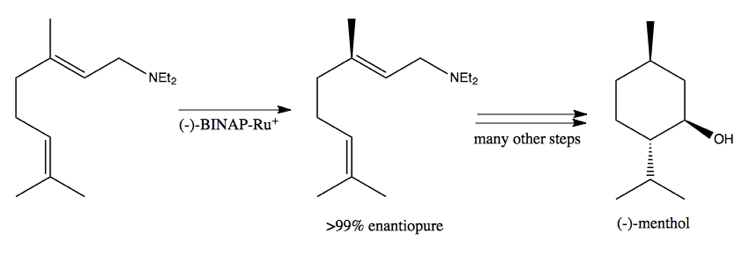
Its phosphenes can bind metal, thus making this a chiral metal catalyst. It can catalyze the following reaction, used in the industrial synthesis of menthol:
Another famous example is trans-cyclooctene. As with the cyclohexane compound diagrammed earlier, the structure at left is ambiguous and can refer to either enantiomer. These two enantiomers do not interconvert, and it is possible to isolate just one or the other.
![]()

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.