Protein folding 06: Amyloid
These are my notes from week 6 of MIT course 7.88j: Protein Folding and Human Disease, held by Dr. Jonathan King on March 12, 2015.
Assignment 5
The readings for this week are:
- Peter Lansbury’s classic paper on the kinetics of amyloid formation for Aβ39 vs. 40 vs. 42 vs. 43 [Jarrett 1993].
- A review by Jeff Kelly and others about the concept of “functional amyloid” [Fowler 2007]
The Aβ paper [Jarrett 1993] studies the solubility of, and kinetics of aggregation of, a variety of synthetic Aβ peptides. Some of the most salient data are:
| peptide | concentration tested | how long it takes to form amyloid |
|---|---|---|
| Aβ1-42 | 20 μM | hours |
| Aβ1-39 | 20 μM | days |
| Aβ26-42 | 200 μM | seconds |
| Aβ26-43 | 200 μM | seconds |
| Aβ26-40 | 200 μM | hours |
| Aβ26-39 | 200 μM | days |
In other words, including residues 40-42 or 40-43 makes it form amyloid much faster, suggesting that the C-terminus of Aβ helps nucleate amyloid formation, and that this nucleation is the rate-limiting step. What’s more, adding even a little bit of one of the “fast” peptides to one of the “slow” peptides makes the latter form amyloid very quickly — again, consistent with the idea that nucleation is what’s rate-limiting.
The review article [Fowler 2007] begins by defining amyloid. Amyloid is often defined operationally, with criteria that Fowler mentions:
- Binds Congo red
- Binds thioflavin T
- Exhibits green-red birefringence
- Has a cross-β X-ray fiber diffraction pattern
But Fowler et al opt for a slightly more specific molecular definition of “a fibrous quaternary structure formed by the assembly of protein or peptide monomers into intermolecularly hydrogen bonded β-sheets”. This still doesn’t help me totally understand what people in the prion field mean when they refer to amyloid vs. non-amyloid PrPSc, [e.g. Rangel 2013]. As an example of amyloid, Fowler offers the NMR structure of Aβ1-42 reported by Roland Riek’s group, with a 4.7Å rung height consistent with measurements from X-ray fiber diffraction [PDB# 2BEG, Luhrs 2005]:
fetch 2beg
hide everything
show cartoon
bg_color white
spectrum count, yellow_red

I was under the impression that high-resolution structures had not been solved for any prion or amyloid other than HET-s. I would be interested if someone could tell me in more detail whether structures such as the one above are considered to represent the “real” in vivo conformation, and if not, why not.
Fowler goes on to review the many reported functional roles of amyloid. Apparently fish and insect eggs are wrapped in an amyloid made of chorion proteins, and the curli protein amyloids in E. coli and other bacteria are thought to help with surface adhesion and perhaps (in the case of pathogenic bacteria) host invasion. These are examples of evolution using amyloid for its structural properties. Then there are examples of epigenetic inheritance, like [PSI+], [URE3], [RNQ+] and HET-s. They go on to talk about fibrin in blood clotting, Pmel17 in apparently sequestering toxic melanin precursors.
Lecture: Aβ and Alzheimer disease
Today’s lecture is organized around five stages of our scientific understanding of Alzheimer disease. At present there are estimated to be 4-5 million Americans with Alzheimer disease.
1. Presentation
For a long time, what we now call Alzheimer disease was simply known as “senile dementia”. Diagnosis was behavioral, based on loss of memory and loss of face recognition in the absence of any loss of motor function.
2. Brain morphology and pathology
Dr. Alois Alzheimer is recognized as having first described a particular neuropathological signature associated with the “senile dementia” package of phenotypes. In the autopsies of the early 1900s, people were cutting relatively thin sections of brains and looking at them with light microscopes. In Alzheimer disease brains there were “plaques” that stained with iodine. Iodine stains starch, so these plaques were referred to as being “starch-like”, which translates into Greek as amyloid. It was later discovered that these plaques also stained with Congo red (which stains starch, too), and that they exhibited birefringence - bending polarized light - which meant they must be made of a fibrous material. The material that was binding the dye was highly insoluble and protease-resistant (proteases were used back then to get rid of proteins so you could study chromatin, or whatever other non-protein molecule you were interested in). When electron microscopy was invented, it was recognized that when these plaques were present, there was also neuronal death.
3. Molecular composition
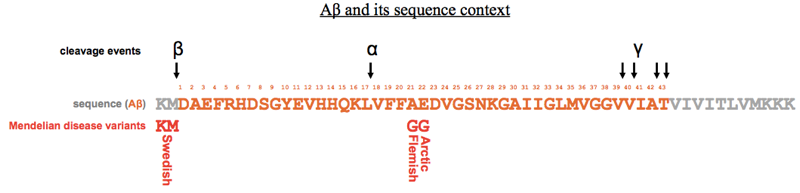
Up until now, the whole literature of Alzheimer disease was written by physicians and pathologists, correlating neuropathological features with behavioral signs. There were almost no biochemists or cell biologists involved in the field. That all changed with the landmark paper sequencing Aβ [Glenner & Wong 1984a]. Glenner and Wong knew from electron microscopy that there were neurofibrillary tangles (NFTs) distinct from the extracellular plaques seen in Alzheimer disease. They used GdnHCl to purify the extracellular plaques, and found that the plaques were composed of a 4.2 kDa peptide they dubbed “beta protein” or amyloid beta or Aβ. They injected the purified protein into rabbits and were able to raise antibodies, which proved to be reactive not only against Aβ but also against a ~700-residue protein they called the “amyloid precursor protein” (APP). Their work led to eventual cloning of the APP gene, and the realization that it was on human chromosome 21, and thus duplicated in Down syndrome, which is associated with early onset Alzheimer disease. They soon found that Down syndrome brains also contained plaques made of Aβ [Glenner & Wong 1984b].
4. Amyloid fibril organization
Subsequent structural studies using X-ray diffraction revealed that Aβ plaques were made of straight, linear fibrils <100nm in length. Dennis Selkoe and colleagues found that the fibrils must have a cross beta structure, meaning that they were composed of beta strands pointed perpendicular to the long axis of the fibril [Kirschner 1986]. They also found that the spacing between these beta strand rungs was 4.7Å.
5. Fibril formation in vitro
Peter Lansbury had studied in one of the first laboratories that worked on solid state synthesis of peptides. He was the first person to figure out how to produce >40-residue peptides at >95% sequence purity. This gave him the ability to study Aβ fibril formation in vitro in a way that no one else could do before, because it was too difficult to obtain sufficiently pure protein from brains, and recombinant protein purification from bacteria had not yet been perfected. Lansbury put synthetic Aβ1-40 into solution, and monitored turbidity as a marker of polymerization. If you were patient enough, you could observe turbidity after several days, and analysis of the resulting material revealed that it was fibrous in nature. He then discovered that if you used Aβ1-42 instead, the turbidity would arise an order of magnitude more quickly. Experiments cross-seeding between the two peptides (and other peptides, as summarized at top) showed that the kinetics of fibril formation involved nucleation and growth — two distinct events, with nucleation as the rate-limiting one. The intellectual and experimental framework for these experiments came from earlier studies of the polymerization of flagella proteins in bacteria.
One example model for how nucleation and growth could differ is as follows. Perhaps monomers very rarely and only transiently misfold. Even more rarely, they laterally associate to form an oligomeric ring with radial symmetry - this is nucleation. Once that ring is formed, it templates additional monomers to add onto its top and/or bottom - this is fibril growth, with the original ring becoming a cross-section of the fibril or perhaps not being preserved in the final structure at all.
The Tycko group did important work identifying different fibril conformations and the possible ways in which different oligomers or “protofibrils” give rise to them [Petkova 2005].
Guest lecture by Peter T. Lansbury, Jr.
Dr. Lansbury’s lecture is entitled Synuclein as a therapeutic target for Parkinson’s disease: 20 years and counting.
Alpha synuclein was first discovered by [Maroteaux 1988]. It turned out to be one of the ten most abundant proteins in the brain, but no one knew what it did or why it was important. At the time, Lansbury was interested in the question of whether one protein could seed or nucleate a different protein to aggregate. Therefore it caught his notice when Tsunao Saitoh reported that a protein he called “NAC” (what we now call synuclein) was involved in Alzheimer disease (as far as I know, this was actually incorrect) [Ueda 1993]. Saitoh was later murdered, so Lansbury felt it was on him to take up the torch of studying this protein [Weinreb 1996]. Soon thereafter, the causal mutation in the Contursi pedigree - a large family with inherited Parkinson disease - was shown to map to the locus of the SNCA gene [Polymeropoulos 1996], and alpha synuclein was shown to be the major component of Lewy bodies, the chief pathological feature of PD [Spillantini 1997]. Lansbury soon discovered by biochemical methods that the A53T substitution of the Contursi pedigree accelerated fibril formation in vitro [Conway 1998].
Lansbury became optimistic that, because PD is quite slowly progressive, drugs that inhibit alpha synuclein aggregation could make a difference in patients’ quality of life even if not administered until symptom onset. The disadvantage, of course, is that it takes a very long time to see whether an already-slow progression has been made even slower by a drug. The prototype model for clinical trials in PD is an 18-month clinical trial of 500 patients, at a price tag of about $200 million, with no success stories in this or any other neurodegenerative disease to inspire investor confidence. This combination of challenges has proven to be a hard sell for some pharma companies.
Lansbury partnered with a fly lab to express alpha synuclein in Drosophila and screen FDA-approved drugs to find compounds that reduced synuclein toxicity measured by an eye phenotype. They found that tocainide was highly effective, but it is a fairly toxic sodium channel blocker. They next asked whether they could find a small molecule that would stabilize a non-aggregating conformer of alpha synuclein. They began with an in vitro screen of FDA-approved drugs, and identified three hits: fluoxetine (Prozac), nortriptyline (Pamelor) and indatraline (all antidepressants). In a secondary screen they found that indatraline and nortriptyline also alleviated the eye phenotypes in the transgenic flies. They then tested some analogues of these in an alpha synuclein mouse model [the mouse model is reported in [Masilah 2000]] and in rat neuronal cell cultures, and got positive results.
Nortriptyline has a terrible side effect though: postural instability. This side effect would be considered unacceptable in Parkinson disease patients. Lansbury and colleagues therefore put this effort on hold for a while. Interestingly, though, a couple of years ago a retrospective study reported that PD patients who happened to take antidepressants had a slower disease progression [Paumier 2012].
Lansbury’s next move was to try to increase autophagy, with the goal of increasing turnover of all soluble proteins including alpha synuclein, theoretically giving it less time to aggregate.
A series of candidate genes studies proposed the UCHL1 S18Y variant as a modifier of Parkinson disease [meta-analysis in Maraganore 2004]. The proposed mechanism was that the protective genotype reduced farnesylation. Lansbury therefore acquired a patent on a farnesyltransferase inhibitor, called CP-609,754 from an oncology company, which had developed this compound for bladder cancer. It had an excellent safety profile but had failed on efficacy in cancer. Lansbury moved a compound, LNK-754, into clinical trials for Alzheimer disease. There were several major problems with the trial. First, the compound causes hypospermia, so they had to recruit only women for the clinical trial. Second, mild-to-moderate Alzheimer disease was the indication, but cerebrospinal fluid biomarkers indicated that many of the recruited patients had been misdiagnosed and did not actually have Alzheimer disease. Third, one woman died of a cerebral hemhorrage - a common complication in Alzheimer disease and probably unrelated to the compound - on day 3 of the phase 1b. Finally, Lansbury gave up and sold the assets to AstraZeneca, which tried to position it for Huntington disease. Sadly, by this time, much of the drug’s patent period had already passed, and the commercial incentive to get it to market was on the decline.
Lansbury next turned his attention back to lysosomes. He became glucocerebrosidase, encoded by the GBA gene. Mutations in GBA cause autosomal recessive Gaucher disease, which can be treated with intravenous recombinant glucocerebrosidase, which Genzyme makes. Gaucher disease is highly comorbid with Parkinson disease - about 10-20% of Gaucher patients eventually get diagnosed with PD (over an 0.5% baseline risk, so an odds ratio of 20-40). Heterozygous carrier parents have increased risk of Parkinson disease, too, with an odds ratio of about 8. (Also, GBA is a significant locus in Parkinson GWAS -EVM). Finally, the causal allele for Gaucher has an allele frequency of about 6% in Ashkenazi Jews, which Lansbury realized makes it possible to gather enough people for a clinical trial by recruiting mutation carriers in just three cities: Tel Aviv, New York, and Boca Raton, FL.
There is cell culture evidence that the relationship between synuclein pathology and reduced glucocerebrosidase activity has bidirectional causality [Mazzulli 2011]. Genzyme therefore took a mouse model of Gaucher and a mouse model of Parkinson disease, and showed that expression of GBA via a viral vector injected into the mouse brain could alleviate synucleinopathy [Sardi 2011, Sardi 2013]. Lansbury and his company, Lysosomal Therapeutics Inc., screened for compounds that could increase glucocerebrosidase activity. They worked from a lead compound, NIH-758, which at 10μM substantially increases glucocerebrosidase activity and decreases total alpha synuclein in iPS-derived neurons from Parkinson disease patients. They were able to obtain analogues that are more potent, have better pharmacokinetics, and are sufficiently chemically distant from the initial lead so as to be patentable. The mechanism of action is still unknown.
Lansbury lists his lessons learned as follows:
- Don’t bother trying to repurpose already FDA-approved drugs, for three reasons. First, they’re unlikely to be highly effective for your indication. Second, paradoxically, many of them actually have terrible safety profiles and would not be approved by FDA today, especially for a chronic, slowly progressive disorder, where you would need to take the drug for years. Third, there’s no way to make money off of them, so it will be hard to raise money for a clincial trial.
- You need the right target.
- You need a wide enough therapeutic window.
- You need a fresh patent with 20 years of life on it in order to maximize commercial potential.
- Glucocerebrosidase activity is appealing as a target because it offers a route to initial approval for Gaucher disease, and then later going after the Parkinson disease market.
Q&A
Q. How does membrane association affect synuclein aggregation?
A. Chris Dobson just published a paper arguing that membrane association nucleates synuclein aggregation [Galvagnion 2015]. However, this is inconsistent with the A30P mutation, which is known to cause Parkinson disease.
Q. How could NIH-758 or its analogues be effective in Gaucher disease if their mechanism of action is to increase glucocerebrosidase activity and those patients lack glucocerebrosidase?
A. Most patients have one of two missense mutations: N370S or L444P. Total loss-of-function mutations are far rarer. In N370S, the protein is translated and localizes correctly, and has some residual enzymatic activity and we would seek to increase that activity. In L444P there appears to be no residual enzymatic activity, so it is less clear whether the compounds would work for individuals with that variant.
Q. Would NIH-758 or analogues work for Parkinson disease patients without GBA mutations?
A. There was a study that found decreased glucocerebrosidase activity in brains of Parkinson disease patients who lacked GBA mutations, so there is some evidence that decreased glucocerebrosidase activity is a more general problem in PD, and therefore these compounds could be broadly effective.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.