Protein folding 09: Sickle cell disease
These are my notes from week 9 of MIT course 7.88j: Protein Folding and Human Disease, held by Dr. Jonathan King on April 9, 2015.
Guest lecture on sickle cell disease by Dr. Kelly M. Knee
Dr. Kelly M. Knee is a research scientist at Pfizer and an expert on sickle cell disease.
Sickle cell disease affects ~70,000 to 100,000 people in the United States, and ~10 million worldwide. The disease was first described by James B. Herrick [Herrick 1910]. The molecular basis was partly elucidated by Linus Pauling and Ibert C. Wells [Pauling 1949], and the exact causal amino acid substitution (E6V) was later identified by V.M. Ingram [Ingram 1956]. This missense variant causes hemoglobin to form long polymers which deform red blood cells into a “sickle” shape. This causes disease by several mechanisms:
- The sickled cells get caught at junctions in capillaries, blocking blood flow and causing inflammation and pain. This is called vaso-occlusive crisis (VOC).
- The sickled cells turn over more quickly and cannot be replaced fast enough, causing anemia
- Red blood cells build up in the spleen, often abolishing its function, causing infections and/or requiring splenectomy
In most developed countries, there are newborn screening programs which allow very early treatment of babies with sickle cell, which dramatically improves outcomes. Globally, infections are the largest cause of death.
The only approved drug for sickle cell is hydroxyurea, which is also used as a chemotherapy for cancer. It is an epigenetic modifier that increases expression of fetal hemoglobin (HbF). HbF is normally about ~5% of circulating hemoglobin, and hydroxyurea can increase this to 10-20%, which reduces sickling. It is not approved for very young children, and some patients cannot tolerate it. In those cases (or when you get a sickle cell crisis in spite of taking hydroxyurea) blood transfusion, palliative pain management (with opiates) and hydration management are the only treatment options.
These limited treatment options don’t help patients all that much. Average life expectancy for people with sickle cell disease in the developed world is still only ~45 years, and in the developing world it is only ~20 years. And quality of life is dramtically reduced.
Red blood cells’ job is to transport O2 throughout the body. To do this, each one contains 270 million hemoglobin molecules and has an O2 concentration of ~5 mM, which is orders of magnitude higher than the concentration of any other protein in any other human cell type.
Hemoglobin is a tetramer, often thought of as a dimer of dimers - two α and two β subunits. See [PDB# 1HBS, Padlan & Love 1985].
fetch 1hbs
bg_color white
hide everything
show cartoon, chain A or chain B or chain C or chain D
color 0xB0171F, chain A or chain C
color 0xFF9912, chain B or chain D
show sticks, organic and (chain A or chain B or chain C or chain D)
color 0x0000CD, organic
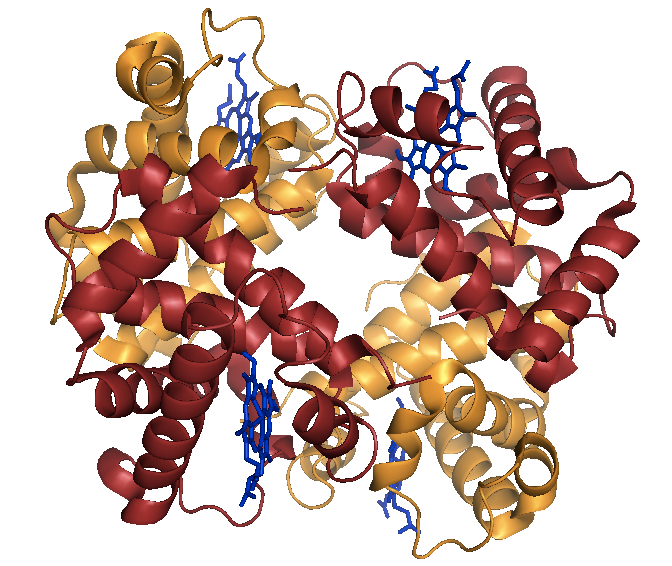
Above: Sickle cell hemoglobin (HbS) in the deoxy state.
Wild-type hemoglobin is referred to as HbA, and sickle cell hemoglobin is referred to as HbS. HbA and HbS are structurally identical in the oxy state and have only minor differences in the deoxy state. Thus, the E6V variant does not disrupt the native function of hemoglobin; instead, the disease is caused by a gain of function.
Switching between oxy and deoxy states of Hb occurs on a femtosecond or picosecond time scale and cannot really be observed in red blood cells. The structural difference between these two states is considerable, involving a 15° rotation of the helices of one subunit with respect to the other. The structural transition is driven, obviously, by the loss of O2.
O2 binding by Hb is cooperative between the four subunits, giving a sigmoidal binding curve, as described here. p50 is a measure of the affinity of Hb for O2. It is considerably lower in HbS individuals than in HbA individuals, because much of the HbS is polymerized.
Polymerization of HbS is driven by exposure of hydrophobic residues. The “EF corner” (of the E helix and F helix), exposed in the deoxy state, includes residues F85 and L88, which form a hydrophobic pocket sometimes called the “EF corner pocket”. This is a very small pocket, and in fact many medicinal chemists object to it being called a “pocket” at all because it is not large enough to be druggable. In HbS, the E6V variant creates a hydrophobic “patch” on the opposing surface. A classical polymerization reaction then ensues, where the V of molecule 1 inserts into the L-F pocket of molecule 2, and the V of molecule 2 inserts into the L-F pocket of molecule 3, and so on. This occurs only in the deoxy state, and when oxygen becomes available again it will partly break up the polymers, but not completely, thus still leaving a polymer seed capable of nucleating more polymerization.
Because the deoxy state is required for polymer formation, sickle cell individuals have worse outcomes when they live at high altitude.
Some drug discovery efforts view Hb, and Hb polymerization, as a target. But for a long time, the fear was that because Hb has a concentration of 5mM, your hypothetical drug would need to be sustained at an unrealistically high concentration in the blood in order to be effective at a 1:1 stoichiometry. New data over the last few years indicate that in fact, substoichiometric drugging of Hb can make a large difference in the propensity to polymerize. Meanwhile, some people are working on other targets for symptomatic treatments - for instance, drugs to reduce sickle cell-induced inflammation. There are also gene therapy and stem cell strategies under investigation.
A single strand of HbS tetramers associates into a 7-strand bundle, and two 7-strand bundles associate to form a 14-strand helix. These bundles pack extremely densely, and eventually cause the red blood cell to rupture.
The kinetics of polymer formation have been modeled in great detail [Ferrone 1985a, Ferrone 1985b, Hofrichter 1986]. The first step is “homogeneous nucleation”, which is association of individual tetramers. This is rate limiting because it is so entropically unfavorable. Once a large enough nucleus (estimated to be about 7 to 10 tetramers) has formed, the energetic gains from covering more hydrophobic patches become overwhelming, and the process speeds up dramatically. There is no good way to study polymerization with a fluorescent readout - for instance, there is no analogue of what thioflavin T does for amyloid. Instead, people study HbS polymerization with turbidity readouts. Incredibly, the polymerization rate is said to be proportional to the concentration of HbS to the fortieth (yes, 40th) power (rate ~ conc40) [Hofrichter 1974, Christoph 2005]. The development of turbidity is rapid at 0.5 mM, very slow at 0.2mM, and not observed at 0.1mM.
All of the phenylalaines in HbS are in uniform environments except for the one in the EF corner pocket (F85). Residue F85’s absorbance depends on how hydrophobic an environment it is in. Therefore, you can use light absorbance to measure the early bundling events (??). Dr. Knee’s PhD work involved using lasers to study the kinetics of HbS polymerization [Knee & Mukerji 2009].
Sickle cell inheritance is autosomal recessive (HBB E6V). Heterozygous carriers do have a small percentage of sickled red blood cells circulating in their blood, but no clinical phenotype. Its shockingly high allele frequency - 4.8% in people of African ancestry [ExAC] - is due to balancing selection, as heterozygous carriers are somewhat resistant to malaria. The molecular basis of why sickle cell heterozygosity inhibits Plasmodium replication is not well understood. The global distribution of HBB E6V (and other hemoglobinopathies) accords almost perfectly with the global distribution of malaria. In some malarial regions, 25-40% of people are carriers. The variant has arisen independently, and undergone balancing selection on separate haplotypes, multiple times in multiple populations.
Given that sickle cell is so common, why hasn’t more progress been made in drug discovery? There are two major reasons. First, Hb was long viewed as an impossible drug target due to its high concentration, as noted above. However, that view has begun to change in recent years. Second, most people with sickle cell live in very poor countries, and even in the developed world, most people with sickle cell have relatively low incomes. However, in recent years both of the both funding agencies and drug companies / biotechs have begun to invest more money in drug discovery for sickle cell.
Here are some past, present and future treatment options that have been / are being explored for sickle cell:
- Allogenic stem cell transplantation has been tested as a therapy for sickle cell patients. It requires an HLA-matched sibling donor. It carries very high risk, with about 10% of patients dying from the procedure.
- Bluebird Bio currently has a clinical trial of delivering lentivirally vectored wild-type HBB [NCT02140554].
- New epigenetic modifiers are under investigation, to increase HbF expression either instead of or in addition to hydroxyurea.
- Small molecule anti-sickling agents that directly target hemoglobin are under development - see the example of 5-HMF below.
- Drugs against other, non-disease-modifying, targets to alleviate symptoms of sickle cell are under investigation. Pfizer just last month reported positive results from a phase 2 trial of rivipansel (GMI-1070; a pan-selectin antagonist) [NCT00911495, Telen 2015] to alleviate vaso-occlusive crisis, and has now advanced this compound to a phase 3 [NCT01119833].
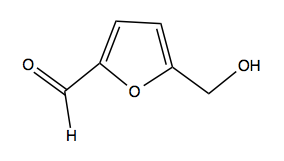
Above: 5-HMF, a candidate anti-sickling drug.
One candidate anti-sickling agent is the covalent drug-like small molecule called 5-hydroxymethyl-2-furaldehyde (5-HMF. above). It is a natural product that can be found in carmelized sugar. It covalently binds the α chain N-terminal amine group, and stabilizes the oxy state of the tetramer. In the oxy state, the β subunit will not dissociate and polymerize. Thus, 5-HMF allosterically inhibits polymerization and sickling. It has two significant liabilities. First is its very high EC50. It binds with 2:1 stoichiometry (as there are two α subunits per tetramer) and you need to bind almost 100% of tetramers in order to have an effect - thus, millimolar concentrations are required for efficacy. In the clinical trial of 5-HMF [NCT01597401], the maximum dose was 4 grams per day, taken in the form of a gross orange juice puree beverage. 5-HMF’s second liability is that it is not very specific - it reacts with amine groups on many other molecules besides hemoglobin. Perhaps secnodary to this, it often causes an immune response. As a result, some researchers have actually become interested in 5-HMF as an immunostimulant for HIV therapy.
5-HMF might never become a drug, but as mentioned earlier, there are encouraging new data suggesting that some anti-sickling agents might prove to be effective disease-modifying treatments without achieving equimolar stoichiometry.
Q&A
Q. Are there genetic modifiers?
A. No protective genetic modifiers have ever been identified for sickle cell. There are some variants that have been suggested to make the disease even worse.
Q. [question involving some confusing analogy to huntingtin]
A. HbS polymerization is entirely unlike huntington or Aβ aggregation, so it is not useful to make that analogy.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.