Protein folding 10: Lessons from virology
These are my notes from week 10 of MIT course 7.88j: Protein Folding and Human Disease, held by Dr. Jonathan King on April 16, 2015.
Virology and protein folding
Some of the foundational work on chaperones came out of the study of DNA viruses, particularly how their coat proteins fold. So today we’ll discuss lessons form virology.
American biomedical research, in many ways, has its roots in the struggle against polio beginning in the 1930s. Polio is transmitted from person to person like a respiratory virus. This is distinct from water-borne infections such as cholera, which rich people had historically avoided by having cleaner water supplies than the poor. The fact that polio was airborne meant no one could escape it, and that is perhaps one reason why research into it was so intensive. Another factor may have been the fact that Franklin Delano Roosevelt was paralyzed by polio. The fundraising effort to fight polio was the origin of the March of Dimes. Some of the earliest efforts to establish cell culture were motivated by a desire to grow poliovirus in culture. Finally, polio motivated a great public health effort to convince people to take the vaccine, which seemed scary because after all, it was made of the virus itself.
Prior to polio, the earliest virology studies had been on plant and bacterial viruses, particularly tobacco mosaic virus, which is easy to isolate in enormous titers by grinding up tobacco leaves, and bacteriophage T4. Delbrück (of Luria-Delbrück fame) studied T4 using funds provided by the March of Dimes.
Mutagenesis of the T4 phage led to the development of genetic map showing the locations of all of the mutations that caused structural abnormalities in the coat:
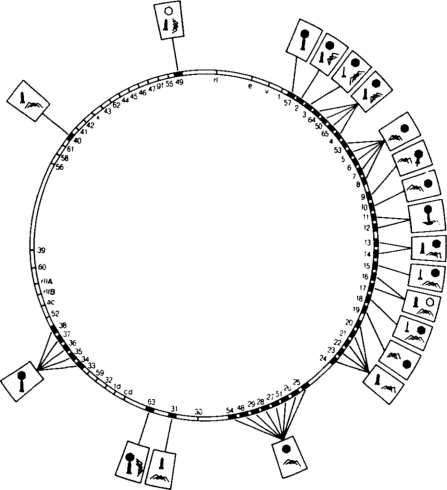
Image from [Aksyuk & Rossmann 2011] via OPENi.
The study of these mutations led to a model for the order in which the head, tail, and tail fibers of the T4 phage were folded and assembled. T4 relies partly, but not wholly, on host chaperonins - the virus actually encodes its own ortholog of GroES that can serve the function of capping the GroEL barrel if the host GroES is unavailable.
Viruses can take a lot of different unique shapes [Baker 1999, see beautiful images in Fig 1]. The structure of icosahedral virus coats was first worked out by [Caspar & Klug 1962], who proposed that each subunit has no symmetry by itself, but that they are arranged in symmetric patterns. The entire virus package, including nucleic acid and coat, is called a virion. Vaccines induce the formation of antibodies against the coat proteins, thus targeting virions. It is only in more recent decades that it has been recognized that it is also possible to target virus-infected cells.
Only a minority of viruses have ever been crystallized and studied using X-ray crystallography. Many more have been studied using cryo-electron microscopy. Cryo-electron microscopy is when you freeze a sample in about 1 millisecond, such that the water forms ice so fast that it does not crystallize - in other words, the H2O molecules freeze in place without reorganizing themselves into an ordered lattice, thus not disrupting the structure of any biomolecule - such as a virus - dissolved in the water. One of the earliest successes in this approach was for adenovirus type 2 [Stewart 1993]. For the herpesvirus capsid, cryo-EM has been used to solve structure down to 8.5Å resolution [Zhou 2000].
Influenza is an RNA virus with 11 different RNA “chromosomes”, a strategy which enables efficient recombination between two different influenza clones that manage to simultaneously infect the same cell.
The most effective HIV drugs act by interfering with folding and maturation of the coat protein. (I couldn’t find a reference for this; the earliest HIV drug, AZT, targeted reverse transcriptase. I did find this table comparing HIV drugs, but it would require some further Googling to find the molecular mechanisms).
Most bacteriophages bind to cell surface polysaccharides or receptors, form a channel and inject their DNA in the cell, leaving their coat outside. (See structures of epsilon15 and P22 [Jiang 2006, Chang 2006]). By contrast, most eukaryotic viruses bind a cell surface receptor, are internalized, and then break out of their vesicle, such that the whole virion, coat and all, is inside the cell. In spite of this diversity, an enormous range of dsDNA viruses - including bacteriophages and eukaryotic viruses, viruses of cyanobacteria and of terrestrial organisms - have coat proteins that share key structural features and are believed to be descended from one common ancestor rather than having arisen via convergent evolution. (ssDNA or RNA viruses, however, are thought to have a separate provenance).
The study of P22 phage infection of Salmonella revealed that ~1h after phage infection, a cell can assemble ~500 new virions with densely packed DNA. In eukaryotic genomes, positively charged histones act to densely pack the DNA, overcoming the replusion due to the negative charge on DNA. In viral capsids, it was hypothesized that the coat proteins must serve the same function. By time lapse it was eventually figured out that the coat proteins indeed assemble into a “procapsid” first. If you run this procapsid’s protein on SDS-PAGE, you find that it contains a lower molecular weight protein that is not found in the mature capsid. It turns out this protein is a scaffolding protein which lends structural integrity to the procapsid, but which is later removed so the capsid can be packed full of DNA. Pulse-chase autoradiography experiments revealed that the synthesis of the coat protein rises and rises after infection of a cell, whereas the scaffold protein is synthesized only transiently. This is because the scaffold molecules are recyclable - after being ejected from a mature capsid ready to be loaded with DNA, they remain intact and recruit newly synthesized coat molecules to form a new procapsid [Botstein 1973, King 1973, King & Casjens 1974].
So P22 uses a conformational / hydrogen bonding mechanism to eject scaffold proteins which are then recyclable. That’s not true of all viruses, though. Many other viruses, including HIV, have proteases which cleave the scaffold proteins in order to release them. Obviously, that means that the scaffold proteins have to be synthesized continuously during virus capsid production, unlike in P22.
If you isolate the coat and scaffold proteins of P22, neither of them will assemble into anything on their own. Only when coat and scaffold proteins are both present will you get assembly [Prevelige 1993]. The initial rate of assembly is proportional to the fifth power of the coat protein concentration and to the second or third power of the scaffold protein concentration. In the assembly of the P22 coat, each time a new monomer binds it is initially in a conformation that makes the coat non-reactive (unable to add new monomers), then there is a conformational change that absorbs the new monomer into the same configuration as the rest of the coat, and now the coat is able to accept yet another monomer [Fuller & King 1980]. Thus, all the conformational change occurs “at the growing edge” - this is also believed to be true in amyloid formation and in basically all polymerization reactions that involve conformational change.
HIV relies on host-encoded prolyl isomerase, cyclophilin A, for maturation of its coat proteins. In fact, host-encoded cyclophilin A is actually incorporated into the virions [Luban 1993, Franke 1994, Thali 1994]. However, there has not been any success in targeting cyclophilin with small molecules as a strategy to inhibit HIV replication.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.