Protein folding term paper: Deciphering the disease mechanism of SOD1 ALS
Below is my term paper for Prof. Jonathan King’s M.I.T. course 7.88j in Spring 2015. If you prefer a paginated, double-spaced version without inline links, see this PDF version instead.
Abstract
The term amyotrophic lateral sclerosis (ALS) refers to a disease phenotype characterized by progressive degeneration of motor neurons, resulting in paralysis and death. Approximately 10% of ALS cases are genetic, and of those, 20% are associated with dominant, mostly missense, mutations in the SOD1 gene. SOD1 encodes a dimeric enzyme, copper/zinc superoxide dismutase, which catalyzes the dismutation of superoxide radicals (O2-), yielding hydrogen peroxide (H2O2). Early reports assumed that SOD1 ALS was caused by a loss of SOD1 protection against oxidative stress, but it soon became clear that causal mutations in SOD1 give rise to a toxic gain of function. The “oxidative damage hypothesis” once held that this gain of function might involve reverse catalysis resulting in generation of oxygen radicals, but this hypothesis has been largely disproven on the basis of in vivo data showing that neither co-expression of wild-type SOD1, nor deletion of a copper chaperone required for SOD1 activity, alter the disease in transgenic SOD1 mutant mice. Instead, it appears that pathogenic mutations predispose SOD1 to aggregate. In vitro data have shown that various SOD1 mutants, compared to wild-type SOD1, have a lower melting point and lower threshold for chemical denaturation or solvent-induced aggregation. Any correlation between these biochemical properties and the clinical features of SOD1 ALS, however, has proven elusive. Nonetheless, the recent demonstration that misfolded states of SOD1 can be transmitted intermolecularly, intercellularly, and to transgenic mice provides compelling evidence that misfolding of SOD1 is indeed a causal event in SOD1 ALS.
SOD1 before SOD1 ALS
Copper/zinc superoxide dismutase, also known as Cu/ZnSOD or SOD1, is a eukaryotic cytosolic enzyme responsible for catalyzing the reaction of superoxide radicals with protons to yield dioxygen and hydrogen peroxide (Figure 1A), thus protecting the cell from oxidative damage [McCord & Fridovich 1969]. Dismutation refers to this simultaneous production of oxidized (dioxygen) and reduced (hydrogen peroxide) products. In humans, SOD1 comprises 153 residues after N-terminal methionine excision and is encoded by the gene SOD1, which is located on chromosome 21 and expressed across all tissues in the body [GTEx Consortium 2013]. SOD1’s abundance in mammalian erythrocytes enabled its early isolation [Mann & Keilin 1938] and determination of its crystal structure (Figure 1B) [Tainer 1982]. Its activity requires that the catalytic site be metallated with one Cu2+ and one Zn2+ ion, coordinated by a total of six histidine residues (Figure 1C) [Parge 1992]. Its ability to prevent the formation of autooxidation products of, for instance, pyrogallol [Marklund & Marklund 1974] enables simple colorimetric assays to measure SOD1 enzymatic activity.

Figure 1. Native function and structure of SOD1. A) SOD1’s native function is to catalyze the dismutation of superoxide (O2-) yielding dioxygen (O2) and hydrogen peroxide (H2O2). B) SOD1 in its native fold is a dimer of Greek key beta barrels with the catalytic site located in the hairpin loops at one end of the barrel (bovine erythrocyte SOD1, PDB# 2SOD). C) The metallated catalytic site visualized in the structure of recombinant human SOD1 (PDB# 1SOS).
Association of the SOD1 gene to ALS
The term amyotrophic lateral sclerosis (ALS) refers to a clinical phenotype resulting from progressive degeneration and death of upper and lower motor neurons, with or without dementia [Boillee 2006]. ALS leads to paralysis and is typically fatal upon denervation of muscles required for respiration [Haverkamp 1995]. ALS is clinically, genetically, and neuropathologically heterogeneous [Al-Chalabi 2012, Renton 2014], with no single protein or pathway implicated in all forms, raising the possibility that ALS may represent a collection of many molecularly distinct diseases [Robberecht & Philips 2013].
Genetic forms of ALS have long been recognized [Myrianthopoulos & Brown 1954] and include dominant and recessive forms collectively comprising ~10% of all cases [Renton 2014]. Linkage studies in the early 1990s established dominant segregation with markers on chromosome 21 in ~20% of genetic ALS families (~2% of total ALS cases) [Siddique 1991], and the causal alleles in these families were soon identified as being a variety of missense mutations the SOD1 gene [Rosen 1993, Deng 1993].
Early reports predicted that these mutations must cause a loss of SOD1 function, with dominant inheritance attributable to haploinsufficiency [Deng 1993]. Over the next five years, however, the evidence in favor of a gain of function became overwhelming:
- Enzymatic activity assays demonstrated that a subset of pathogenic SOD1 mutants retain wild-type levels of activity [Borchelt 1994, Hayward 2002].
- Genetic ablation of Sod1 in mice, even in a homozygous state, is reasonably well-tolerated, with reduced neuronal survival following axonal injury but no degeneration or paralysis phenotype reminiscent of SOD1-linked ALS [Reaume 1996].
- Overexpression of mutant SOD1 in transgenic mice causes motor neuron loss, paralysis, and death in a dose-dependent fashion, with accelerated disease onset in high-expressing lines (Figure 2A) [Wong 1995].
- Disease in these transgenic overexpressers is unaltered by co-expression of endogenous Sod1 [Bruijn 1998].
In addition, all 31 of the ALS-associated SOD1 mutations reported to date in the ClinVar database [Landrum 2014] are missense, with the exception of one nonsense mutation (L126X) located in the final 20% of the coding sequence, where protein truncation may not necessarily result in a total loss of function [MacArthur 2012]. Alone, any one of these lines of evidence is imperfect - for instance, in vitro enzymatic activity does not guarantee that correct folding occurs in vivo, and gene inactivation in mice sometimes results in a far milder phenotype than the corresponding human disease [Chamberlain 2007]. Collectively, however, these findings point strongly towards a gain of function.
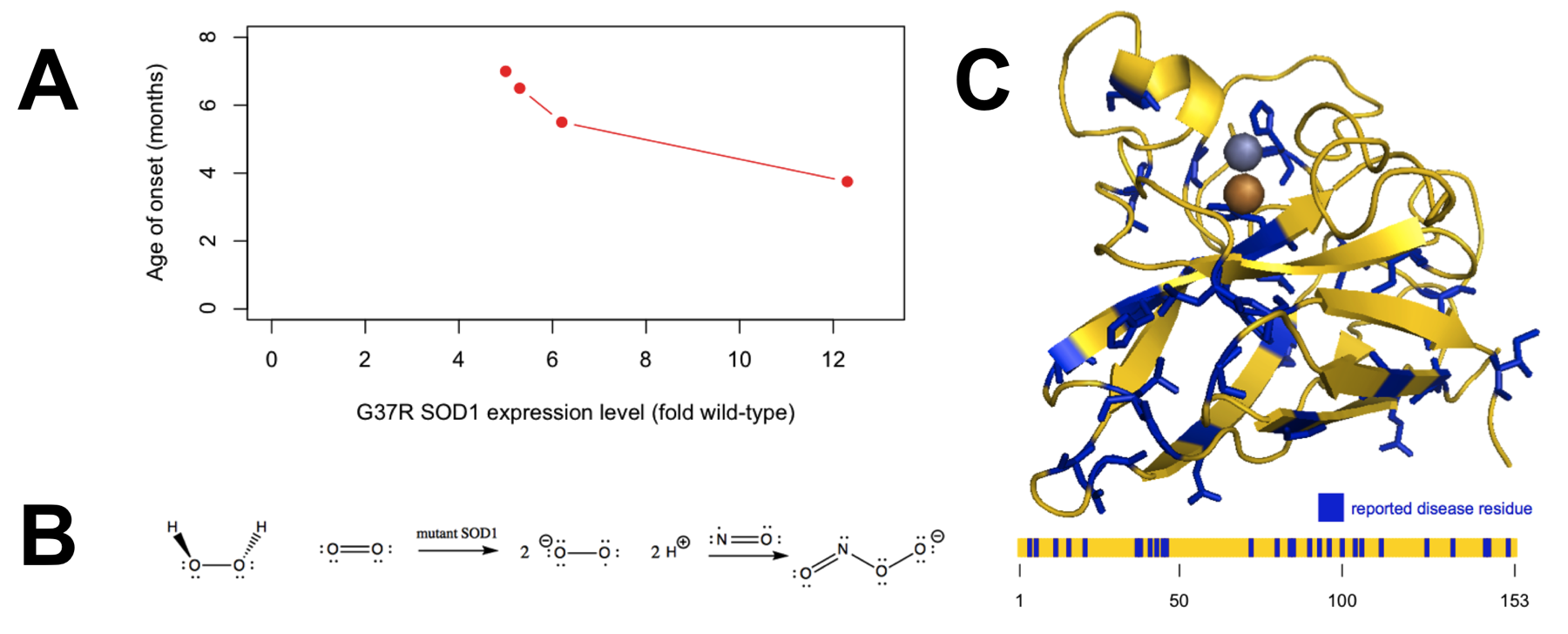
Figure 2. Evidence regarding the mechanism by which SOD1 causes disease. A) Overexpression of mutant SOD1 causes disease in a dose-dependent manner, indicating a gain of function. Plotted data are from Table 1 of [Wong 1995]. B) The oxidative damage hypothesis proposed that mutant SOD1 catalyzed the production of superoxide from hydrogen peroxide, ultimately yielding peroxynitrite. C) The distribution of the 31 reportedly pathogenic SOD1 mutations collected in the ClinVar database (ncbi.nlm.nih.gov/clinvar, accessed April 30, 2015) appears random with regard to position in the three-dimensional structure (top) and primary structure (bottom).
The oxidative damage hypothesis
By the late 1990s, most - though not all [Saccon 2013] - investigators had become convinced that SOD1 mutations cause ALS by a gain of function. Amidst many proposals for the identity of the gained toxic function [Ilieva 2009], two major schools of thought emerged [Cleveland & Liu 2000]. The oxidative damage hypothesis held that pathogenic mutations conferred upon SOD1 an increased affinity for hydrogen peroxide, and thus caused it to catalyze the production, rather than dismutation, of toxic radicals (Figure 2B). The aggregation hypothesis held that the mutations caused SOD1 to misfold and that some resultant oligomeric or aggregated species of the protein was neurotoxic.
The position of reported pathogenic mutations within the protein (Figure 2C) appears fairly random, with no particular preference for the catalytic site, and according to ClinVar, only one of the six metal-coordinating histidines is the site of a disease-causing missense mutation (H46R). In addition, different SOD1 missense mutations vary greatly in both their level of enzymatic activity and their ability to bind metal ions, which are presumed to be necessary both for normal enzymatic activity and for the putative reverse catalysis supposed by the oxidative damage hypothesis [Valentine & Hart 2003]. All of these observations make the oxidative damage hypothesis seem unlikely a priori.
Nonetheless, the oxidative damage hypothesis gained currency in the mid- to late 1990s. One early study compared the ability of wild-type versus G93A mutant SOD1 purified from Sf9 cells to perform reverse catalysis in vitro, producing superoxide radicals from hydrogen peroxide [Yim 1996]. This was measured by performing electron paramagnetic resonance (EPR) on the small molecule 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a spin trap, to detect radical adducts produced by reaction of DMPO with nascent superoxide. The mutant enzyme was reported to produce a slightly higher amount of DMPO radical adducts, and by varying the concentration of H2O2, they estimated its affinity (Km) for H2O2 to be 25.8 mM, versus 34.5 mM for wild-type SOD1, at pH 7.6. Thus, it was hypothesized that in vivo, the mutant either binds H2O2, or stays bound to nascent H2O2, and generates damaging oxygen radicals. It is unclear how this difference in Km could be responsible for a toxic gain of function in vivo, however, given that the physiological concentration of H2O2 is estimated at ~50μM [Gautam 2006], about three orders of magnitude lower than these Km values.
Another study used a nearly identical in vitro paradigm with EPR and DMPO, studying wild-type and two mutant SOD1s, G93A and A4V, purified from yeast [Wiedau-Pazos 1996]. Again, the experiments were performed at a supraphysiological 30 mM concentration of H2O2. The mutants were reported to produce greater amounts of DMPO radical adducts, and the production of these adducts by wild-type or mutant SOD1 appeared to require that SOD1 be metallated with copper, as the signal could be abolished by addition of copper chelators DDC or penicillamine. These chelators were also reported to reduce apoptosis associated with overexpression of mutant SOD1 in the rat neural cell line CSM14.1, supposedly establishing the in vivo relevance of the findings. A third study reported that depletion of zinc from either mutant or wild-type SOD1 rendered the enzyme toxic to rat primary neuronal cultures, and that this toxicity could be rescued by nitric oxide synthase (NO) inhibitors, thus suggesting that the toxicity was mediated by production of superoxide leading to peroxynitrite formation (Figure 2B). The authors went on to suggest that perhaps mutant SOD1’s toxicity might therefore be mediated by a deficiency in zinc binding.
All of this indirect evidence for the oxidative damage hypothesis from in vitro and cell culture studies was surprising in light of the regional distribution of pathogenic mutations (Figure 2C) and, moreover, was at odds with available in vivo data [Cleveland & Liu 2000]. The disease observed in transgenic mice overexpressing mutant SOD1 is unaltered by co-expression of wild-type SOD1 [Bruijn 1998], whereas if the toxic gain of function were mediated by production of superoxide, one would expect disease to be attenuated by the availability of some wild-type enzyme to dismutate these superoxide radicals.
Fortuitously, a new in vivo test of the oxidative damage hypothesis soon became possible. A yeast mutagenesis screen identified a chaperone required for loading copper into SOD1 [Culotta 1997]. Dubbed copper chaperone for SOD1 (CCS), the chaperone proved to have orthologs throughout eukaryota (in humans, the gene is CCS). Ccs knockout mice exhibit only ~15% of wild-type SOD1 activity levels in their spinal cord, suggesting that the chaperone is largely essential for SOD1 function in vivo [Wong 2000]. Assuming that copper is required not only for normal SOD1 enzymatic activity but also for the putative reverse activity of mutant SOD1, then under the oxidative damage hypothesis, Ccs knockout should dramatically attenuate the toxic effects of mutant SOD1 expression. When three different mutant SOD1 transgenic mouse lines were crossed to Ccs knockout background, however, no difference in disease course was observed [Subramaniam 2002].
The aggregation hypothesis
The results of the Ccs knockout experiment helped to shift focus towards the aggregation hypothesis. Initial interest in this hypothesis probably arose from a neuropathological observation: SOD1 ALS patients have ubiquitinated aggregates of SOD1 in the neurons and astrocytes of their spinal cord [Kato 2000]. This feature is recapitulated in G93A SOD1 mice, and indeed, lower-order multimers can be detected months before large aggregates appear [Johnston 2000].
Throughout the early 2000s, a series of studies examined the in vitro unfolding and aggregation properties of cherry-picked SOD1 mutants. By this time, proper metallation of bacterially expressed SOD1 could be achieved by co-expression of CCS [Lindberg 2002]. Investigators use term holo to refer to metallated SOD1, and apo to refer to SOD1 depleted of metal, usually by chelation with EDTA.
When equilibrium unfolding was measured by circular dichroism, apo SOD1 was found to unfold at lower GdnHCl or urea concentrations than holo SOD1, and in either state, most of the mutants studied (A4V, C6F, D90A, G93A, and G93C) unfolded at lower denaturant concentrations than the corresponding wild-type protein [Lindberg 2002]. Similarly, when melting points were measured using differential scanning calorimetry, apo SOD1 melted at a lower temperature than holo SOD1, and in either state, the studied mutants (A4V, G93A, G93R, and E100G) melted at lower temperatures than the corresponding wild-type [Stathopulos & Rumfeldt 2003]. When the kinetics of unfolding were measured in 4M or 6M GdnHCl, these mutants also unfolded more rapidly than wild-type [Stathopulos & Rumfeldt 2003]. When aggregation was induced by addition of trifluoroethanol (TFE) and measured by thioflavin T fluorescence, the mutants also aggregated at lower TFE concentrations than wild-type (10-12% vs. ~15 vol/vol TFE respectively) [Stathopulos & Rumfeldt 2003]. Across these mutants, the concentration of TFE required to induce aggregation was correlated with the measured melting point [Stathopulos & Rumfeldt 2003]. Similarly, in heat-induced aggregation measured by right-angle light scattering, an indicator of turbidity, the mutants aggregated at lower temperatures than wild-type [Stathopulos & Rumfeldt 2003].
One in vitro study dispensed with mutants altogether, instead subjecting purified wild-type SOD1 to metal-catalyzed oxidation by CuCl2 and measuring unfolding using 8-anilinonaphthalene-1-sulfonic acid (ANS), a small molecule which fluoresces when bound to exposed hydrophobic patches of proteins [Rakhit 2004]. When SOD1 underwent metal-catalyzed oxidation, it was found prone to aggregate at lower protein concentrations. Meanwhile, dynamic light scattering and analytical ultracentrifugation indicated that the oxidized protein was present at least partly as a monomer rather than the native dimer. This led the authors to hypothesize that SOD1 monomers may represent one intermediate on the pathway to aggregation, and that mutant SOD1 has a lower energy barrier to dissociating into monomers [Rakhit 2004].
While all of these results are certainly consistent with the aggregation hypothesis, the use of GdnHCl, heat, or TFE in these studies is arguably no more physiologically relevant than the addition of 50 mM hydrogen peroxide in the oxidative damage studies performed years earlier. Proving that mutant SOD1’s misfolding properties are actually relevant to disease in vivo is difficult. Several investigators have asked whether biochemical properties of the various disease-associated SOD1 mutants correlate with clinical parameters observed in patients with these mutations, such as age of onset or disease duration. Frustratingly, however, most such studies have found no correlation [Ratovitski 1999, Prudencio 2009, Vassall & Stubbs 2011]. The only major study to report a positive result in this area focused primarily on predicted, rather than empirically measured, properties of the mutant proteins [Wang 2008].
An orthogonal approach to demonstrating the causality of SOD1 misfolding in disease would be to show that the disease can be transmitted by misfolded SOD1. After all, successful transmission experiments have become central lines of evidence in favor of the prion hypothesis for other neurodegenerative diseases [Prusiner 2012, Jucker & Walker 2013]. This has become an active area of investigation for SOD1 over the past several years, and evidence so far indicates that intermolecular and intercellular transmission of SOD1 can be observed experimentally, with some evidence for in vivo transmissibility as well.
In one study, mutant H46R SOD1 was purified from Sf9 cells and induced to aggregate by the addition of 20% TFE [Munch 2011]. When these aggregates were labeled with Dylight 649, a red dye, and added to culture media of mouse N2a neuroblastoma cells, intracellular red puncta could be observed within one hour, indicating that cells are capable of taking up extracellular SOD1 aggregates. When N2a cells were stably transfected to express H46R SOD1 fused to GFP, the GFP appeared diffuse throughout cytosol at baseline. Upon addition of the aforementioned TFE-induced aggregates to culture media, the GFP signal formed green puncta co-localizing with the red puncta from exogenous aggregates. This demonstrated that aggregates taken up from the extracellular space could template aggregation of endogenous mutant SOD1.
Another study demonstrated that mutant SOD1 can also template the misfolding of wild-type human SOD1 [Grad 2011]. That study utilized a 4-bp frameshift insertion [Andersen 1997] properly described as p.G127fs5X but often referred to in the literature as G127X. The neoepitope created by the frameshift allows discrimination of mutant and wild-type protein. When HEK cells were transiently transfected to express the frameshifted protein, it became possible to detect the endogenous wild-type SOD1 using antibodies to disease-specific epitopes that are inaccessible in properly folded SOD1 and are also absent from the mutant protein, thus demonstrating intermolecular transmission of a misfolded state to the wild-type protein [Grad 2011]. As further evidence for intermolecular transmission, the wild-type protein in these experiments also formed non-native intermolecular disulfide bonds [Grad 2011], as has been observed in mouse models of SOD1 ALS [Jonsson 2004, Jonsson 2006, Furukawa 2006, Karch 2009]. Moreover, once wild-type SOD1 misfolding has been induced in this manner, it can be transmitted to untransfected cells by transfer of culture media over at least five serial passages [Grad 2014].
These experiments collectively demonstrate that SOD1 misfolding is transmissible between molecules and between cells, consistent with a prion mechanism in SOD1 ALS. Yet the observation that misfolded aggregates can transmit does not yet demonstrate that they are what cause the disease. Accordingly, some cell culture studies have sought to demonstrate that SOD1 misfolding is cytotoxic. For instance, one study showed reduced survival of cells that form aggregates upon transfection with mutant protein, and suggested that sequestration of the proteasome by aggregates may be responsible [Matsumoto 2005]. It is difficult, however, to extrapolate from the death of transiently transfected cells in culture to causes of motor neuron death in vivo.
To date, only one study has examined the transmissibility of SOD1 ALS in mice [Ayers 2014]. The study focused on inoculation of spinal cord homogenates into recipient mice heterozygous for a transgene array expressing G85R SOD1 fused to YFP. This genetic background was expected to confer heightened susceptibility to SOD1 aggregates, as these mice do not become spontaneously sick, but mice homozygous for this transgene array do become sick, and mice expressing both G85R SOD1-YFP and G93A SOD1 become sick more rapidly than mice expressing G93A SOD1 alone.
When these G85R SOD1-YFP mice were injected with spinal cord homogenates from spontaneously sick, paralyzed G93A SOD1 mice, some of the recipients became paralyzed within a median of ~5 months, though the attack rate was not 100%. Upon serial passage - injecting spinal cord homogenates of paralyzed G85R SOD1-YFP recipient mice into a new round of G85R SOD1-YFP mice - the attack rate rose to 100% and the median incubation time dropped below 3 months. Paralysis was not observed in any G85R SOD1-YFP mice injected with PBS (to at least 14.4 months) or with spinal cord homogenates from wild-type mice (to at least 9.9 months) as controls. The G93A spinal cord homogenates did not transmit disease to wild-type mice nor to mice over-expressing wild-type SOD1, nor did they accelerate disease in G93A mice. While these non-transmission results suggest that misfolded SOD1 may exhibit only limited in vivo transmissibility, they also serve as controls to establish that the transmission of disease to G85R SOD1-YFP mice likely resulted from templated misfolding of endogenously expressed mutant SOD1, rather than from any direct toxicity of the G93A homogenates.
Conclusion
The transmission studies reviewed here establish only a limited capability for misfolded SOD1 to transmit its conformation and the associated disease state. For instance, it has not been demonstrated that misfolded SOD1 can transmit to wild-type animals, nor that titers of infectivity are maintained over many serial passages, as shown for PrP prions. Nevertheless, even the level of transmissibility demonstrated here probably constitutes the best evidence so far that the misfolding of SOD1 is indeed the cause of SOD1 ALS. It remains unclear exactly which species (for instance, oligomer or aggregate) of SOD1 is neurotoxic, and by what mechanism it kills neurons. In spite of this limitation, there may be therapeutic promise in generic strategies to prevent the propagation of misfolding. For instance, some investigators have searched for molecules to prevent SOD1 dimers from dissociating into monomers [Wright 2013], and antisense oligonucleotides to reduce expression of SOD1 have shown promise in a transgenic rat model [Smith 2006] and appear to be well-tolerated upon intrathecal delivery in SOD1 ALS patients [Miller 2013]. It is to be hoped that further elucidation of the mechanisms of SOD1 misfolding and neurotoxicity will reveal additional therapeutic avenues.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.