Chemical biology tutorial 2: cheminformatics and small-molecule activity databases
These are my notes from tutorial 2 in Harvard’s Chemistry 101: Chemical Biology Towards Precision Medicine course, taught by Dr. Paul Clemons on October 29, 2015.
How computers view molecules
Similarity is relative. Quantifying similarity between different molecules requires a digital representation of the molecules and a formula for quantifying difference or similarity. For some of the earliest examples of computer representations of molecules, see [Randic 1979, Carhart 1985]. More recent representations include SMILES and InChI. SMILES has difficulty representing tautomers, isotopes, and a few other edge cases, so InChI is more comprehensive, but also newer (circa 2004) so there is not as much legacy support for it. ChemDraw literally uses digital representations of the drawing, in inches, rather than of the underlying structure: it stores coordinates and identities of atoms, and then a match table of atoms that are bonded to one another, along with stereochemical information for each bond.
Tanimoto distance only measures the distance between bit strings, it doesn’t imply anything about what information is encoded in the bit string. You can generate bit strings (or “fingerprints”) representing various different sets of features of molecules. A currently popular model for bit strings is extended-connectivity fingerprints (ECFP) of various diameters (for instance, ECFP4 or ECFP6) [Rogers & Hahn 2010].
Assay databases
ChemBank was a project from ca. 1998-2008, capturing data from about 4,700 screens covering a total of 1.3 million unique structures. It is no longer being maintained. Now several alternatives are available. We’ll discuss NIH’s BARD, which is the direct successor of ChemBank and holds all the data from the MLPCN projects, but several other databases have overlapping goals, data, and capabilities: for instance NCBI’s PubChem, UCSD’s BindingDB, UCSF’s ZINC, and EBI’s ChEMBL. Collectively, these databases offer the promise of allowing one to computationally query the biological activity of compounds, rather than relying on predicted chemical properties.
As one example of how to use these resources, a phenotypic screen found a molecule that induced insulin expression in pancreatic alpha cells, and they then queried ChemBank to identify 13 molecules with similar bioactivity profiles, two of which were annotated as kinase inhibitors [Fomina-Yadlin 2010]. The original hit was subsequently confirmed to be a kinase inhibitor as well.
When you look at a molecule in BARD, among other things it will display a figure for ‘Assays - Active vs. Tested’:
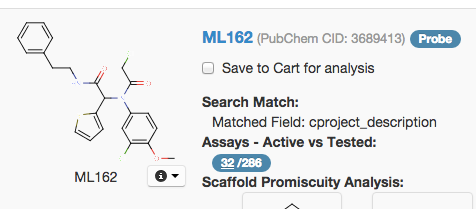
In this example, it is 32/286. But that doesn’t mean the 32 are independent. BARD includes all assays, not just primary screens, so in many cases, a large fraction of the Active assays will be re-tests and secondary and counterscreens after finding the original hit, meaning that hits which do validate (this example, ML162, is now recognized as a probe) often appear to have high hit rates. If you want to know whether the molecule is a pan-assay interference compound, the Scaffold Promiscuity Analysis plug-in is more useful than just the overall active vs. tested ratio.
As you browse, you can add both compounds and assays to your Cart. Once you have everything you are interested in, you can click ‘Visualize’ in the Cart to generate a spreadsheet of which compounds were active in which of your assays of interest.
The ‘Bio-Activity Summary’ tells you everything you need to know about one molecule’s performance in various assays — a table with assay details and the compound’s EC50 and full dose-response curves.
The ‘Linked Hierarchy Visualization’ uses structured assay definitions across a variety of types of annotation to let you explore your compound’s activity. So for instance you can see if your compound is only hitting in luciferase-based assays, which might suggest it is just a luciferase inhibitor.
A useful tool for exploring protein-protein interaction data is STRING. See also STITCH.
A more wikipedia-like interface to learning more about probes - their advantages and limitations, which negative controls to use, etc. - is ChemicalProbes.org.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.