What do we know about ApoE?
I’m confused about ApoE. And the more I read, the more I think I’m not alone.
Of all the known genetic risk factors for late-onset Alzheimer disease, APOE genotype has by far the largest effect size — a single E4 allele increases your risk by ~3X [Harold 2009]. If you believe, as I do, that human genetics is a valuable source of validation for therapeutic strategies, then it follows that ApoE might be considered as one possible drug target for preventing Alzheimer disease.
The trouble is, no one seems to know the mechanism by which the E4 allele increases Alzheimer risk. There is not even any agreement on whether E4 represents a gain of function or a loss of function, and therefore on whether one should strive to develop inhibitors or activators of ApoE. Accordingly, people have taken approaches that are all over the map. Some think ApoE is beneficial, and have sought to therapeutically upregulate APOE expression [US Patent US6274603B1, Jiang 2008, Cramer 2012]. Others view ApoE as the enemy, and look to its knockdown or its inhibition with monoclonal antibodies as potential therapies [Kim 2011, Kim 2012, Bien-Ly 2012, Choi 2015]. Still others believe that the answer is isoform-dependent, and that one should seek to downregulate the risk-increasing E4 isoform and/or upregulate the protective E2 isoform [Hudry 2013, Hu 2015].
It amazes me that there can be so little agreement on such a fundamental issue. And it’s not for lack of interest. One of the studies mentioned above [Cramer 2012], which claimed that bexarotene upregulated Apoe and in turn reduced Aβ plaque burden in a mouse model of Alzheimer’s, quickly came under fire when four academic groups failed to replicate it, and it recently made the news again when Amgen announced that it had also failed to replicate it.
Upon turning to the literature, the first problem that I found is that APOE is too well-studied, with 21,301 PubMed hits as of today. Paradoxically, all this literature makes my task harder, rather than easier. The literature is so crowded with different claims about APOE that it becomes incredibly difficult to tell what is real. For instance, in a quick Google Scholar search I spotted more than ten candidate gene studies proposing an association between APOE genotype and either the risk, age of onset, or phenotype of prion disease, even though in the largest prion disease GWAS to date [Mead 2012], there was not so much as a whiff of association at the APOE locus. To believe everything in the literature, it seems, would be to believe this one protein is found in every subcellular compartment, interacts with every other protein, modulates every mouse phenotype, and is a risk factor for every neurological disease. I soon developed the following postulate: for every hypothesis that you can imagine about ApoE, there exists a paper that claims it’s true.
To try to sift through this morass, I figured I’d start with what I know best: human genetics. That’s not to say that human genetics papers about APOE are any more likely to be correct than papers about mouse models or cell biology or anything else, but rather, human genetics is a body of literature that I know how to read. I don’t always know a good mouse from a bad mouse or a representative image from a cherry-picked one, but I have at least a decent handle on the methods and statistical thresholds that pass for evidence in human genetics. So the question of this post, then: what does the human genetic data tell us about APOE? Be warned, even on this subject I’m far from an expert, so this post is very likely to contain mistakes and omissions.
First, some general facts about APOE variation in humans
APOE consists of 3 exons located on chromosome 19. In terms of numbering, the coding sequence is 318 codons long including the stop codon, but the first 18 codons encode a signal peptide, so the numbering scheme in common use in the literature subtracts 18 from the codon number. Two variants are common worldwide, C130R (often called C112R) and R176C (often R158C), and another variant, R163C (or R145C) has a frequency of 6% in Africans. And of course, as with any gene, there are loads of other less common variants — a total of 99 different protein-altering variants in the current ExAC release. Other than the variants discussed above, plus one variant in the signal peptide (N14K), none of these has an allele frequency greater than 1% in any large continental population. Note that, as is convention, the amino acid codes above are given in comparison to the reference genome, but in the case of C112R, R is actually the ancestral allele, shared by chimpanzees and mice alike. This means that 112C, which is the reference and the most common allele in all major world populations, actually arose in humans and swept to its current 70-90% allele frequency before the out-of-Africa bottleneck. The R158C variant, which is found in cis with 112C, must therefore have arisen later on that same new haplotype.
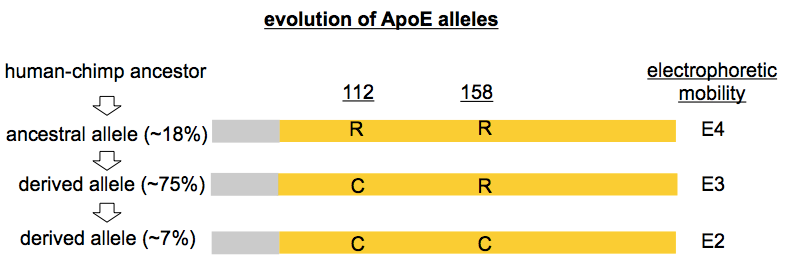
People don’t often refer directly to these missense variants by their codon and resulting amino acid substitition as I’ve done above. Instead, one often hears a specific nomenclature for APOE variants, one which, as far as I can tell, arose by historical accident. ApoE protein was studied using biochemical techniques years before DNA sequencing was in common use, and it was noticed early on that different versions of the protein had different isoelectric points and could be separated on 2D gels. (I’m not sure of the earliest citation for this, but for an example gel see [Moriyama 1992]). The term “E3” came to refer to the electrophoretic mobility of the protein encoded by the most common allele (now the reference). “E2” referred to any ApoE protein with one fewer positive charge, and “E4” referred to any ApoE protein with one more positive charge. It turns out that most of the time, the underlying missense variants that gave rise to these differences were R158C (E2) and C112R (E4), though there were exceptions: that common African R145C variant mentioned above also has “E2” electrophoretic mobility, and there were other, rarer, variants as well, such as “E2 Fukuoka” (R224Q) [Moriyama 1996]. As an additional historical curiosity, “E1” referred to ApoE protein with two fewer positive charges than the reference E3 allele, and reported E1 alleles included K146E [Moriyama 1992] as well as G127D cis R158C [Weisgraber 1984]. Nowadays, when people say E2 or E4, they’re no longer thinking of electrophoretic mobility, but instead are simply referring to R158C and C112R respectively — and for the avoidance of confusion, I will refer directly to the missense variants in the rest of this post, with parenthetical reference to the “E” term in common use.
Blood lipids and cardiovascular disease risk
Years before APOE was ever associated with Alzheimer disease, its protein product was known as a component of lipoprotein particles in the bloodstream. As far as I could find, the earliest reference to what we now call ApoE may be the characterization of an “arginine-rich” (34 arginines, we now know) protein enriched within very low density lipoproteins (VLDL) particles in the blood of patients with a disorder called dysbetalipoproteinemia [Havel & Kane 1973]. Within a few years, the arginine-rich protein, christened ApoE, was known to be present not just in VLDL, but also in chylomicrons and HDL [reviewed in Smith 1978].
Fast-forwarding to modern day, we know that APOE genotype is associated with blood lipids and cardiovascular risk in a few different ways. The APOE locus is reproducibly associated with LDL cholesterol levels in genome-wide association studies [Willer 2008, Teslovich 2010, Global Lipids Genetics Consortium 2013]. None of those GWAS papers discussed the direction of effect nor whether the locus was also associated with coronary artery disease, but those issues were examined in detail in an enormous meta-analysis (~100,000 individuals) [Bennet 2007]. That study found that R158C (E2) resulted in reduced LDL and reduced risk of coronary artery disease, while C112R (E4) increased both. For LDL, R158C homozygotes (E2/E2) had 41 mg/dL lower LDL than C112R homozygotes (E4/E4). In an allelic model, compared to the reference genome (E3), R158C (E2) was associated with reduced risk of coronary artery disease (odds ratio = 0.80) while C112R (E4) was associated with increased risk (OR = 1.06).
At first glance, then, this all sounds straightforward: R158C (E2) is good, and C112R (E4) is bad. It’s not quite so simple, though. Although R158C (E2) on average is associated with reduced risk of cardiovascular disease, there seems to be a lot of variance in people’s outcomes, and the earliest studies of E2 actually examined its role in causing disease. It’s confusing, and I only wrapped my mind around it after finding one good thorough review [Utermann 1987]. The R158C (E2) variant is described as being defective in receptor binding, thus causing ApoE protein, which is found on chylomicrons, VLDL, IDL, HDL, to stay in the bloodstream and circulate longer. R158C (E2) is associated with increased levels of circulating ApoE (associated with one or more of chylomicrons, VLDL, IDL, HDL) and decreased levels of circulating ApoB (associated with LDL). So although LDL is apparently always decreased in these individuals, which is good, VLDL and other lipoprotein particles can be increased, sometimes pathologically so, such that a subset of R158C homozygotes (E2/E2) actually suffer from a disorder called either dysbetalipoproteinemia or type 3 hyperlipoproteinemia. These people have very high total cholesterol, and develop xanthomas (visible deposits of cholesterol under their skin) and early-onset heart attacks. I couldn’t find any reference for what proportion of R158C homozygotes (E2/E2) develop this condition, but it must be fairly small, since in the aggregate, even in a homozygous state, this variant appears to be associated with reduced risk of coronary artery disease [Bennet 2007].
To further add to the confusion, APOE loss-of-function variants also cause a Mendelian, recessive form of dysbetalipoproteinemia. Individuals lacking a functional copy of APOE have low LDL but high levels of all other lipoprotein particle types, especially VLDL, which is increased by several-fold. This, at first, seemed incomprehensible to me: how could the absence of ApoE cause the same disorder as homozygosity for R158C (E2), which is associated with increased circulating ApoE? Utermann speculates that any deficiency in ApoE binding to its receptors, whether due to R158C or due to total lack of ApoE, results in upregulation of the LDL receptor (thus reducing LDL) but an inability to scavenge “remnant” lipoprotein particles such as VLDL and IDL (and thus an increase in levels of those particles). To date, several families with this recessive disease of APOE deficiency have been reported [Ghiselli 1981, Mabuchi 1989, Kurosaka 1991, Lohse 1992, Feussner 1996, Mak 2014], with casual variants including a variety of nonsense, frameshift, and splice variants. There was also one individual with an “E1” missense allele in trans to a frameshift [Feussner 1992]. Following the discovery of these families, Apoe knockout mice [Zhang 1992] became a widely used mouse model of atherosclerosis.
Is the APOE loss-of-function disease purely recessive, or is there a heterozygote phenotype? No one seems to know for sure. Certainly, the severe phenotypes of xanthomas and early onset atherosclerosis were only reported in double null individuals, and not in their heterozygote relatives. One proband did have an affected father, but he was deceased and his genotype was unknown, and 3 out of 6 of his children were affected [Ghiselli 1981], so he may well have been a homozygote himself. Some of the papers cited above do present VLDL, LDL, triglyceride and other blood lipid levels for one or two heterozygote relatives, but none of their numbers look to be glaringly outside the range in the general population, so it is not clear whether they are significantly shifted from the population distribution. One of the papers [Feussner 1996] phenotyped 10 heterozygotes, two of whom were even deemed to be affected, but even then, none of the lipid levels were significantly different in hets from controls after multiple testing correction. Figuring out for sure if there is a heterozygote phenotype will take a larger N that we don’t have yet (see discussion at bottom).
Alzheimer disease
Almost two decades after the association of ApoE to blood lipid phenotypes began to be understood, people started to notice that ApoE protein could be immunodetected in Aβ plaques in Alzheimer disease brains [Namba 1991]. This made APOE an attractive target for candidate gene studies, and within a couple of years, it was reported that APOEgenotype was associated with the risk of Alzheimer disease [Strittmatter 1993, Corder 1993].
The amazing thing was that this was actually true. A rare success story of the candidate gene era, the association between APOE and Alzheimer disease has been resoundingly validated time and again, and modern-day GWAS have shown that APOE genotype actually has far and away the largest effect size of any common genetic variant on Alzheimer disease risk [Naj 2011, Hollingworth 2011, Lambert 2013].
Compared to the reference (E3) allele, R158C (E2) is protective, while C112R (E4) confers increased risk. I’ve seen a wide range of estimates of what exactly the odds ratios are [Corder 1994, Tsai 1994, Genin 2011]. The largest meta-analysis I found [Farrer 1997] has it as follows:
| genotype | odds ratio |
|---|---|
| E2/E2 | 0.6 |
| E2/E3 | 0.6 |
| E3/E3 | 1.0 |
| E2/E4 | 2.6 |
| E3/E4 | 3.2 |
| E4/E4 | 14.9 |
Odds ratios are a funny thing. In prion disease, which is very rare, even an odds ratio of 100 can mean a clinically negligible amount of risk [Minikel 2016]. But for Alzheimer disease, which is so common by old age a priori, some of these odds ratios verge on Mendelian: C112R (E4) homozygotes exhibit >50% penetrance of Alzheimer disease by age 85 [Genin 2011].
Other phenotypes
To see if there were any genome-wide significant associations of other traits with APOE, I turned to the NHGRI/EBI GWAS catalog and ran a couple of queries:
wget http://www.ebi.ac.uk/gwas/api/search/downloads/full
cat full | grep APOE | awk -v FS="\t" -v OFS="\t" '$29 > 7.3 {print $2, $8, $29}' | wc -l
cat full | grep APOE | awk -v FS="\t" -v OFS="\t" '$29 > 7.3 {print $8}' | sort | uniq -cAs of today, there are a total of 111 published studies reporting genome-wide significant (P < 5×10-8) associations at the APOE locus, nominally pertaining to 35 different traits. On closer examination, though, it seems that Alzheimer disease and blood lipids are the only two real underlying traits here. Most of the 35 GWAS traits are either synonymous or closely correlated with one of those two traits. For instance, reported associations with Aβ levels and verbal declarative memory simply reflect the association with Alzheimer disease, and reported associations with metabolic syndrome, C-reactive protein, and blood metabolite ratios are presumably secondary to the effects on blood cholesterol. Age-related macular degeneration (AMD) also makes the list of associated traits, but here too, there is speculation that this may simply reflect an association of blood lipid profiles with AMD [McKay 2011]. Similarly, because heart attacks and Alzheimer disease are both major killers, it seems reasonable to suppose that the recently reported association of APOE with longevity [Fortney 2015] results from one or both of these factors.
Are the blood lipid and Alzheimer associations related?
Above, I’ve suggested that all of the other APOE-associated traits are simply secondary to APOE’s associations with blood lipids and with Alzheimer disease. If so, then the question naturally arises, what about these two traits? Does APOE genotype affect the two traits by two separate mechanisms, or is the Alzheimer association simply a result of changes in blood lipid composition?
A link seemed unlikely a priori, because C112R (E4) has such a modest effect on blood lipids, with only an odds ratio of 1.06 for coronary artery disease, which makes it hard to believe that the odds ratio of >3 for Alzheimer disease is secondary to that. But I had a look anyway, and from what I see in the literature, there doesn’t seem to be any evidence at all for a mechanistic link between the two. If there were, then one would expect that other genes and environmental factors that affect blood lipids would affect Alzheimer disease risk, and that doesn’t seem to be the case.
For one, blood lipids themselves have never been shown to be predictive for Alzheimer disease. This argument can be further extended to environmental risk factors and to cholesterol-lowering drugs. Diet has been linked to blood cholesterol levels for decades [Keys 1965], yet has never been convincingly linked to Alzheimer disease risk, no matter how great the proliferation of Alzheimer’s prevention cookbooks on Amazon. And even though millions of millions of people take statins to lower their LDL levels, the claims that statins reduce Alzheimer risk [Jick 2000] remain unproven and controversial [Zandi 2005]. As Stanley Prusiner put it, when asked by an audience member at Prion2013 what people could do to reduce their risk of dementia, “It’s not how many crossword puzzles you do. It’s called luck.” As of today, scientists have not confidently identified a single environmental risk factor for Alzheimer disease, even as hypercholesterolemia is increasingly well-understood and well-treated.
I also looked for evidence of shared genetics. At last count, there were 19 loci associated with Alzheimer disease risk [Lambert 2013] and 157 loci associated with blood lipid phenotypes [Global Lipids Genetics Consortium 2013], and APOE is the single point of overlap between these two lists. To double-check this conclusion, I turned again to the GWAS catalog, and ran a quick query in R:
gwas = read.table('full',header=TRUE,sep='\t',quote='',comment.char='')
dim(gwas)
# [1] 25217 34
gwas$alzheimer = grepl('Alzheimer',gwas$DISEASE.TRAIT)
gwas$lipid = grepl('holesterol',gwas$DISEASE.TRAIT)
cholesterol_genes = unique(gwas$REPORTED.GENE.S.[gwas$lipid & gwas$PVALUE_MLOG > 7.3])
alzheimer_genes = unique(gwas$REPORTED.GENE.S.[gwas$alzheimer & gwas$PVALUE_MLOG > 7.3])
alzheimer_genes[alzheimer_genes %in% cholesterol_genes]
# [1] NA "APOE" "APOE, TOMM40" "APOC1" "intergenic" "APOE, TOMM40, PVRL2"Sure enough, even accounting for every study in the GWAS catalog, APOE is still the only genomic locus associated with anything related to Alzheimer’s and anything related to cholesterol. (TOMM40, APOC1, and PVRL2 are neighboring genes that fell within APOE’s linkage peak in some early studies).
So it is not broadly the case that genetic variants that affect blood lipids also affect Alzheimer risk — only APOE does. None of this is to rule out the possibility that the effects of APOE genotype on lipids and on Alzheimer disease risk are somehow mechanistically linked, but if there is a link, it is incredibly specific to APOE — it’s certainly not as simple as “blood cholesterol affects the risk of Alzheimer disease.”
Does APOE E4 increase Alzheimer risk by a gain or loss of function?
I argued above that the evidence concerning ApoE’s mechanistic link to cholesterol and cardiovascular disease is complicated and confusing. R158C (E2) is associated with an increased, but still low, risk of a specific blood lipid disorder, dysbetalipoproteinemia, while still overall being associated with reduced LDL and reduced cardiovascular disease risk. C112R (E4) is associated with increased LDL and increased cardiovascular disease risk. APOE loss-of-function homozygotes have low LDL but wildly increased VLDL and an extremely high risk of early onset heart attacks.
But if we do not think that there is necessarily any mechanistic link between the effects of APOE on blood lipids and the effects of APOE on Alzheimer disease risk, then all this conflicting evidence may actually have no bearing on the question at hand.
Instead, maybe we should ask a separate question: do individuals with APOE loss-of-function variants (whether heterozygous or homozygous) have an increased or decreased risk of Alzheimer disease compared to the general population?
I would have thought someone would have asked this question by now, but I don’t see any papers on it. Perhaps it’s just because we don’t have the data yet. The handful of humans who are double null for APOE (in the studies cited above) have very high cholesterol that apparently is hard to manage with available drugs [Mak 2014], so sadly, it’s not clear how many of them have lived long enough that they would have developed Alzheimer’s, even if their risk were high. Only a small number of heterozygote relatives were phenotyped in those studies, and it is not at all clear how detailed a family history was collected, encompassing how many individuals, and whether a family history of dementia would have been noticed and written up if it were present — particularly since some of the early studies pre-date the discovery of APOE’s association with Alzheimer’s.
Meanwhile, APOE loss-of-function variants are rare enough in the population that a genotype-first approach may not yet be well-powered either as of 2016. In all of ExAC there are only 12 individuals with nominally protein-truncating variants, and most affect non-canonical transcripts or occur in the final exon, where a total loss of function is not guaranteed.
I see a few potential roads forward. If extended families of the already-identified APOE null individuals are keen to participate in research, one could imagine researchers following the alleles out far enough in the family tree to find a large number of heterozygotes. Or, in a few years when there are larger cohorts of individuals willing to be re-contacted — maybe the million person cohort — some researcher will manage to re-contact enough unrelated APOE LoF hets and ask them whether there is dementia in their family history. Alternately, case-control cohorts may grow large enough that a significant enrichment or depletion of LoF alleles among Alzheimer disease cases may become apparent.
A paper a few years ago proposes the idea of small molecule “structure correctors”, ligands that make a C112R (E4) allele fold more like a reference (E3) allele [Chen 2012]. The approach is intruiguing for being totally mechanism-agnostic: we don’t know why E3 is better, but it is, so let’s make the protein act that way. But finding a ligand that does exactly this thing is a much narrower target to hit than simply finding a ligand. So until we have a definitive answer on the gain vs. loss of function issue, the road forward is difficult indeed.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.