Presymptomatic imaging
In a number of different dementias, brain imaging is believed to offer important predictive power for whether people will develop disease or will progress towards more severe disease. For example, MRI in presymptomatic people at risk for Huntington’s disease is thought to disclose changes in regional brain volumes perhaps 15 years before clinical onset [Paulsen 2008, Paulsen 2010, Aylward 2011]. In Alzheimer’s, PET scans using the amyloid imaging agent Pittsburgh compound B are being used as clinical trial enrollment criteria [Sevigny 2016], and are thought to have some predictive value for whether cognitively normal adults will develop Alzheimer’s disease [Morris 2009].
Do we have any hope of leveraging these sorts of presymptomatic imaging biomarkers for clinical trials in prion disease? Prion disease differs from these other dementias in being exceptionally rapid. The median time from first symptom to death is only 5 months in sporadic CJD (the most common subtype of prion disease), and about 1 year in some genetic subtypes [Pocchiari 2004]. And “first symptom” here is being assessed retrospectively by interviews with family members. The time from diagnosis to death is even shorter, because on average, about two-thirds of the disease course is spent just reaching a diagnosis [Paterson 2012]. Compare this to Huntington’s disease, frontotemporal dementia or Alzheimer’s, where the typical disease duration is more on the order of a decade or two [Foroud 1999, Roberson 2005]. Because prion disease progresses very rapidly after first symptom, one might worry that the disease process leading to that first symptom may also move quickly, leaving only a rather narrow window for presymptomatic detection.
At the symptomatic stage, it’s well established that diffusion-weighted MRI is a good clinical tool for diagnosing sporadic prion disease and particularly for distinguishing it from other rapidly progressive dementias [Shiga 2004, Vitali 2011]. Some genetic subtypes of prion disease are less obvious on MRI [Vitali 2011], but other imaging techniques can be useful — for instance, FDG-PET has revealed hypometabolism in the thalamus of individuals symptomatic for genetic prion disease with the D178N cis-129M haplotype [Perani 1993].
Can any of these techniques be used to identify changes in the brain before the onset of symptoms? One systematic way to find out is to recruit asymptomatic people at risk of genetic prion disease to a longitudinal study and image them serially for years to see if any changes can be detected preceding onset. It’s a long road to walk: age of onset is incredibly variable in genetic prion disease — for instance, the age of onset of people who have fallen ill with E200K genetic prion disease has a standard deviation of 10 years [Minikel 2014] — so a researcher may wait a very long time to see even a few people become symptomatic. Michael Geschwind’s Early Diagnosis of Human Prion Disease study at UCSF, and Simon Mead’s study at University College London have each been doing this for over a decade, but neither has reported any presymptomatic imaging changes yet.
So far, all of the data we have on potential presymptomatic imaging changes comes from five separate N of 1 case reports. The latest of these, published earlier this week [Zanusso 2016], prompted me to go back and take a look at all of them. One report is from a longitudinal study, while four are from apparently serendipitous imaging scans undertaken for various reasons on individuals who later developed prion disease. A challenge for me is that I can’t read these images myself at all. MRI and PET scan images mean nothing to me: in these figures there is usually an arrow pointing to some region of interest, and even when I squint at it, I cannot begin to discern what’s interesting about it. So I am basically taking these researchers’ word as to what they found in the images. If any neurologists or radiologists who’ve had a look at the data in these papers care to comment, I would love to hear other opinions on what the images show.
Here is a summary of those case reports:
- Daniela Perani and Pietro Cortelli have been longitudinally following the original D178N cis-129M family in the Veneto region of Italy since 1993. At the start of the study, they enrolled 9 people with the mutation, at ages ranging from 25 to 65, and seem to have imaged them with 18FDG-PET roughly every 2 or 3 years. 18FDG is simply a radiolabeled sugar which can reveal relative rates of energy usage in different brain regions. Of the four individuals who became symptomatic in the first 13 years of the study, one had undergone a PET scan 13 months prior to disease onset, which was read as being slightly abnormal, with a 16% decrease in thalamic metabolism intensity [Cortelli 2006]. This is milder than after disease onset — the same patient had a 40% decrease in the thalamus by 7 months post-onset. They didn’t find presymptomatic imaging changes in any of the other three patients who became symptomatic, but none of them had undergone scans as close to onset as 13 months.
- One case report describes a 68 year old man who requested brain MRI because his sister had died of a subarachnoid hemorrhage. By diffusion-weighted MRI he was found to have a slight hyperintensity on the right side of the cortex [Satoh 2011]. This hyperintensity worsened one month later on follow-up, and about three months after the initial scan he had difficutlies with his right hand, which quickly evolved into rapidly progressive dementia. He ended up having autopsy-confirmed sporadic prion disease.
- In one case report, a 68 year old woman who presented with a headache and nausea was found to have some changes consistent with prion disease by diffusion-weighted MRI. She returned to the clinic a few months later and was diagnosed with prion disease based on further MRI studies, cognitive decline, and the presence of the PRNP V180I variant [Terasawa 2012]. This was reported as presymptomatic detection, which it may well be, although reading it, I don’t quite feel 100% confident that it was truly presymptomatic. It is unusual but not unheard-of for things like headache or dizziness to be reported as the first symptom in prion disease [Brown 1986, Rabinovici 2006]. Also, the individual had not died as of when the paper was published, and without autopsy, the diagnosis of prion disease is not definite either. The V180I variant appears to confer only a low, perhaps 1%, lifetime risk of prion disease [Minikel 2016], so it is not inconceivable that this individual was actually in the early stages of some other, more common form of dementia.
- A 65 year old woman underwent MRI for a chronic headache that was diagnosed as being the result of a cortical stroke. Diffusion-weighted MRI showed cortical hyperintensity in the right cortex, but she lived healthily for another 12 months before developing rapidly progressive dementia. A total of 15 months after the initial scan, she was diagnosed with prion disease based on worsened MRI findings, EEG, and the presence of tau, 14-3-3, and RT-QuIC prion seeding activity in the CSF [Verde 2016].
- A 73 year old woman was undergoing annual MRI scans by way of follow-up for a tumor, and was eventually diagnosed with prion disease on the basis of further diffusion-weighted MRI, EEG, as well as RT-QuIC seeding activity in nasal brushings and RT-QuIC seeding activity, tau, and 14-3-3 in CSF [Zanusso 2016]. The diagnosis was later confirmed by autopsy. None of her MRIs had initially been read as indicative of prion disease, but in hindsight, the diffusion-weighted MRI scans taken 1 month and 14 months prior to onset were read as showing hyperintensity in the cortex, while a scan 26 months prior to onset still seemed normal even with the benefit of hindsight.
As far as I know, these five are the only such reports in the literature right now. If you know of others, please point me in the right direction.
Here are a few reflections. It’s not clear how well, if at all, a prediction of prion disease could be made prospectively on the basis of any of the imaging results reported here. Some of these papers specifically state that presymptomatic imaging was only indicative of prion disease in hindsight, while others don’t address the issue. No one seems to have made the claim that they were able to read the presymptomatic images and predict that the patient was about to develop prion disease. Also, note that because most of these reports are from serendipitous clinical visits rather than longitudinal studies, there is presumably some publication bias at work here. It is possible that other individuals who have died of prion disease had also undergone MRI or PET scans within a year prior to onset, but if those scans were negative for prion disease-related changes even in hindsight, then such results would probably be less likely to be published than the positive results in these papers. The FDG-PET study [Cortelli 2006] did report on three patients in whom no changes were found prior to onset, and it would be interesting to know how many more examples like this are out there unreported.
With those caveats, plotting the data from these five reports as a timeline does show some commonality:
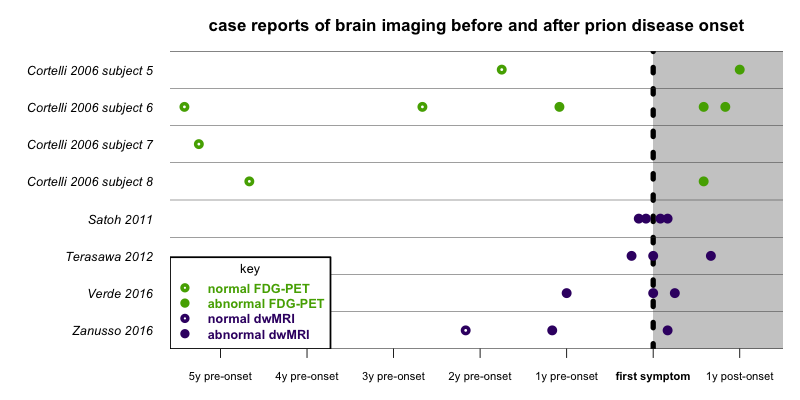
This plot shows that the limited data available from these eight patients in five case reports are pretty consistent. All scans at ≥21 months before onset were normal, and all scans at ≤14 months before onset were abornmal. This is not a large sample size, but all the signs so far point to the first imaging-detectable changes in the brain happening a year or two before onset, and no earlier. This is not nearly as much “runway” as one has before symptom onset with, say, MRI in Huntington’s disease. While a year or so before onset is not necessarily too late, the ideal time to intervene therapeutically when we have a drug for prion disease is probably even earlier.
There is no doubt that longitudinal studies of imaging in presymptomatic people are important. At the same time, all of the evidence so far makes me wonder whether biochemical biomarkers may be able to provide a much earlier diagnosis than imaging in prion disease. Evidence from animal models is suggestive in this regard. In mice infected with ME7 prions, diffusion-weighted MRI revealed changes only at 126 days post-infection (dpi), well after the earliest behavioral symptoms (burrowing changes were apparently observed at 84 dpi) and only slightly before the beginning of terminal-stage disease (described as being at 133 dpi) [Broom 2007]. In contrast, in hamsters intracerebrally infected with prions by various routes, prion seeds were detectable by RT-QuIC in both brain and CSF at least as early as 30 dpi, in animals that don’t develop clinical signs until 90 dpi, and die around 120 dpi [Orru 2012]. Because RT-QuIC is highly sensitive and specific on both CSF and nasal brushings in symptomatic prion disease patients [Atarashi 2011, McGuire 2012, Orru 2014, Orru 2015], it will be important to determine how early before onset, if at all, prion seeds can be detected in presymptomatic people at risk for genetic prion disease.
Update: Here is one more presymptomatic imaging paper, on E200K carriers from Israel: [Cohen 2015]. The findings are similar to the papers reviewed above.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.