Food and Drug Law 01: Introduction to FDA
These are my notes from class meeting 1 of Harvard Law School’s Food and Drug Law course, led by Prof. Peter Barton Hutt on January 3, 2017. Reading for today’s class meeting is chapter 1 (pages 1-25) of Food and Drug Law 4th Ed..
Today’s readings and course provided an introduction to the the U.S. Food and Drug Administration, including its history, regulatory scope, and its structure and function.
Key events in FDA’s history and prehistory
Historical precedent for regulation of food safety extends back at least five millennia [Hutt 1984]. Clay tablets concerning food and drug regulation have been discovered dating from ~3000 BCE in Ancient Sumeria. One Sumerian statute said that any innkeeper who charged for a false quantity of ale would have her hand cut off. In ancient times, there were herbs, spices, and animal products that were considered as drugs — in fact, pepper was among the most oft-prescribed medicines in Ancient Rome. The fact that these early “medicines” were foods is probably the original reason why food and drug regulation are still unified under one regulatory statute and agency. Ancient Roman laws banning stellionatus (fraud) prohibited selling adulterated or spoiled food, and Pliny the Elder’s The Natural History discussed a wide variety of drugs and cosmetics in use at the time, often warning of widespread adulteration. Until ca. 1950, the term adulteration simply meant mixing an expensive substance with a cheap one (e.g. milk with water, pepper with juniper berries).
In Medieval England, merchants importing black pepper would gargle with it to ensure quality and throw out batches that were adulterated with charcoal. This process became known as garbling, and gave rise to the modern words garbage and garble (citation needed). A 1263 English statute prohibited selling any food “not wholesome for man’s body”, a standard remarkably similar to FDA’s current standard prohibiting adulterated food as that which “contains any poisonous or deleterious substance which may render it injurious to health”. Medieval England had both civil and criminal penalties for selling unsafe or impure foods, and trade guilds played a major role in cracking down on such offenses, by expelling members who violated standards.
Despite all this precedent, there was no regulatory paradigm anywhere in the world for experimental testing of safety until the FDA came along. In the 1500s, Paracelsus said that the only difference between a poison and a remedy is the dose, a concept today known by such catch phrases as “the dose makes the poison” or “there are no toxins, only toxic quantities”. But there was no concept of animal testing in Paracelsus’s time, so the only way to determine a poisonous dose was from observing humans. Moreover, most of the ancient and medieval precedents were concerned with adulteration as an economic issue — say, scamming customers by charging milk prices for a product that was only half milk and half water — rather than a health issue.
A turning point in the evolution towards modern food and drug regulation came with the advent of chemistry in the 1800s. German chemist Friedrich Accum’s Treatise on Adulteration of Food, published in 1820, seems to be the earliest recorded observation that food adulteration was not only an economic issue, but also a health one — for instance, adulterated milk might be mixed with impure water that contained pathogens that could cause disease. Highly publicized tragedies soon caused this view to become widespread and to become a bedrock motivation for regulation. In the 1858 Bradford Sweets incident in Britain, a confectioner making peppermint candies bought sugar from a corrupt supplier who was adulterating sugar with gypsum, and who in turn had bought gypsum from a corrupt supplier who was selling gypsum contaminated with arsenic. 20 children died and hundreds were sickened after eating the arsenic-laden candies. This eventually led to Britain’s Pharmacy Act 1868, which was possibly the first food and drug legislation explicitly concerned with safety (although it used the term poison; the term safety was not applied to food and drugs until decades later).
Meanwhile, chemical techniques made it possible to test for the presence of food adulterants, and over the 1800s the United States iteratively established mechanisms for chemical testing of foods, which by 1901 had taken the form of the USDA Bureau of Chemistry [Hutt 1990], the bureau which would eventually become the FDA. (Incredibly, by 1902 the Bureau had a “poison squad” of 12 people who ingested different chemicals to determine their safety). Chemistry wasn’t the only innovation of this era. In parallel, the invention of vaccines, starting with the smallpox vaccine in 1796, almost immediately led to the sale of fraudulent vaccines, which in turn eventually led to regulation of biologicals. And Benjamin Waterhouse, a co-founder of Harvard Medical School, by testing the smallpox vaccine on his own children, created some of the earliest precedent for organized human experimentation.
In addition to technological advances, regulation also grew out of justified public healthc concerns, as life expectancy was on the decline in American cities in the 1800s. Lemuel Shattuck’s 1850 report on behalf of the Massachusetts Sanitation Committee named food and drug adulteration, along with sewage, as one of the major reasons why the life expectancy was dropping, and helped to spur interest in regulation. The states passed various laws to regulate specific foods, biologicals, and drugs over the 1800s, including the smallpox vaccine, imported drugs, tea, and so on, and there were a few Congressional actions too, but most regulation was by the states, and there was no generalized scheme for regulation of food or drugs. Because urbanization was occurring and trade was on the rise, consumers and producers were increasingly separated from each other, and public concern about food and drug adulteration grew, with specific scandals spurring change. For instance, in the 1901 St. Louis Tragedy, 13 children died of tetanus after receiving a tainted batch of diphtheria antitoxin (a biological to this day produced from horse serum), Congress passed the Biologics Act in 1902. Upton Sinclair’s The Jungle, which exposed repulsive meat-packing practices, was what finally precipitated the passage of the Food and Drugs Act of 1906, which gave the USDA Bureau of Chemistry the power to regulate food and drugs that were traded in interstate commerce.
The 1906 law was still incredibly loose compared to the regulatory environment we know today. For instance, pharmacies could still sell narcotics over the counter, as long as the packaging was accurately labeled. Indeed, a 1917 report complained that the law lacked “any restriction whatever upon the use of many of the most virulent poisons in drugs.” Procedurally speaking, one of the most profound differences versus today was postmarket enforcement (the 1906 law only provided for action against non-compliant products already on the market) as opposed to requiring premarket approval. Nonetheless, the regulators empowered by the 1906 law were very aggressive about bringing criminal charges against corrupt producers, and did a remarkable amount to clean up the food and drug supply in the U.S. They undertook about 2,000 seizures per year (compared to ~20 per year in the FDA today).
Over subsequent decades the Bureau of Chemistry became the FDA, and there were various calls for expansions of FDA’s powers. Another national tragedy eventually helped to spur change: in 1937, more than a hundred people died after taking a liquid formulation of the antibiotic sulfanilamide dissolved in the poisonous antifreeze diethylene glycol (see “Elixir Sulfanilamide”). The following year, Congress passed the Food, Drug, and Cosmetic Act (FD&C) of 1938. This law brought cosmetics and medical devices under FDA’s purview, expanded FDA’s authority to regulate labeling (though not advertising, which fell to the Federal Trade Commission), established the first affirmative labeling requirements, banned poisonous ingredients, and heightened FDA’s enforcement powers. Procedurally, the FD&C changed drug regulation from a system of postmarket enforcement to one of “premarket notification”, with manufacturers required to demonstrate safety — but still not efficacy — before selling their product. The system was that drug manufacturers were required to provide FDA with 60 days’ advance notice of a new product, and FDA could either deny the application, or if FDA did nothing, the product could be marketed by default (hence the term “premarket notification”). The FD&C remains the bedrock legal basis for FDA’s existence and powers, and basically every piece of legislation concerning FDA since then has simply amended the FD&C.
The next major expansion of FDA’s power occurred in 1962, with the so-called Drug Amendments. Procedurally, these replaced the earlier system of “premarket notification” with a system of “premarket approval” where affirmative approval was required. And more substantively, these amendments tasked FDA with regulating not only the safety, but also the efficacy, of drugs, and also gave FDA the authority to regulate clinical trials. Together, these changes meant that FDA now governed the entire process of what drug makers needed to prove, and how they needed to prove it, in order to bring a drug to market. A useful history of the 1938 and 1962 legislation is found in [Merrill 1996].
To date, Congress has amended the FD&C Act over two hundred times, most recently with the 21st Century Cures Act. The majority of these >200 amendments have heightened FDA’s regulatory power and expanded the scope of what it regulates. There have been a few notable exceptions, where Congress has restricted FDA’s power: for instance, the Vitamin-Mineral Amendments of 1976 are the reason that FDA does not have much authority over vitamins and dietary supplements. But overall, FDA’s regulatory power has steadily expanded, and this trend has persisted regardless of which political party holds power. FDA has come to regulate food, drugs, cosmetics, devices, animal feed and drugs, tobacco, and things that emit radiation: it even has a Memorandum of Understanding with the Federal Aviation Administration to jointly regulate laser light shows to make sure they don’t blind pilots. In some cases, the expansion of FDA’s power hasn’t come from new regulation, but from FDA taking over roles previously played by other agencies — for instance, it took over the regulation of biologics, which since the Biologics Act of 1902 had been carried out by the National Institutes of Health. All told, FDA is now responsible for regulating products that comprise about 25% of U.S. GDP.
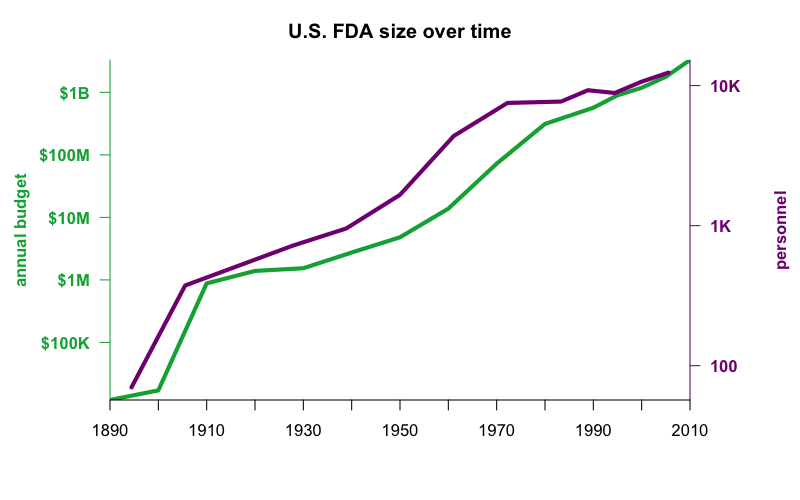
Above: the FDA budget and number of personnel over time, data from p. 25 of Hutt, Merrill, and Grossman, 2014, Food and Drug Law 4th Ed. Latest data point is 2010, with $3.29 billion and 12,381 employees.
How FDA works
Legally speaking, the Department of Health and Human Services is responsible for implementing the FD&C Act, and FDA is the division within HHS that does this. Originally, the Secretary of HHS appointed the FDA Commissioner, but Congress amended FD&C in 1988 such that the Commissioner is appointed by the President and approved by the Senate. This means that, whereas some other federal regulatory agencies are supposed to be “independent regulatory commissions” (Securities and Exchange Commission, Federal Trade Commission, etc.), the FDA is explicitly not independent. In principle, FDA could therefore be subjected to political pressure more easily than many other agencies, but our textbook argues that this is not the case in practice. Instead, FDA’s public visibility and clear scientific basis for its decisions help to insulate it from political maneuvering. In addition, only its top-ranking officials are political appointees: Commissioner, Deputy Commissioner, Associate Commissioners and sometimes Chief Counsel (a position that Prof. Hutt once held). The rest of FDA’s employees are career feds heavily insulated from political changes, and they are the ones who make almost all of the decisions.
Indeed, FDA is described as being unique in just how bottom-up it is. FDA has an enormous regulatory scope, and the Commissioner cannot feasibly even be aware of, let alone have a hand in, any but a tiny fraction of its decisions. The >100 Congressional bills that have amended FD&C usually only provide direction in broad strokes; the details of how FDA will interpret Congress’s will appear in the form of Guidances, of which there were over 2,000 in 2004-2014 alone, and these are drafted by career employees, not political appointees. And beyond Guidances, much of FDA’s regulatory discretion and authority lies in the individual decisions of whether or not to approve a given drug (or other product), and here too, it is the FDA employees, not leadership, that decide.
All that being said, FDA is also described as being more closely watched and more often questioned by Congress, the media, and the public, than perhaps any other regulatory agency. FDA comes before Congress for an average of about 30 oversight hearings per year.
FDA has its headquarters in Hyattsville, MD, home of the Commissioner and six “Centers” corresponding to six types of products that FDA regulates: drugs, biologics, food, devices, veterinary products, and tobacco. The Center for Drug Evaluation and Research (CDER) is the center responsible for drugs, including new approvals. FDA also has dozens of field offices around the country (and abroad) mostly concerned with enforcement and inspection, and runs the National Center for Toxicological Research in Jefferson, AR.
FDA receives an appropriation from Congress but also, since 1992 and on an ever-increasing basis, gets its funding from “user fees” paid by the commercial entities whose products are being regulated.
Miscellaneous
- Under the Freedom Of Information Act, all of the safety and efficacy data submitted by drug sponsors are considered confidential and cannot be released under FOIA rules until after the drug is approved.
- Food and drug law has a weird numbering system: §402 of the FD&C Act is §342 of the U.S. Code. To translate from FD&C numbering to USC numbering, always delete the middle zero and prepend a three. Food and drug law can be browsed as Title 21 of U.S. Code.
- Although the >200 amendments have dramatically changed regulation, many early cases still form foundations of food and drug law. For instance, the definition of “safety” still in use dates from a 1914 decision.
- Prof. Hutt marks thalidomide as the moment when FDA shifted its focus from mostly food to mostly drugs.
- 1976 was the first time that the Supreme Court ruled that the First Amendment applied to commercial speech. Since then, FDA has lost 13 consecutive First Amendment cases in the courts. Fundamentally, FDA regulates both substances (e.g. food, drugs) and words (claims about those substances), and it is currently grappling with how to regulate the latter while complying with the First Amendment.
Covington & Burlington issues “e-alerts” with summaries of new legislation affecting FDA, here are a few relatively recent ones for reference:
- 2007: FDAAA
- 2012: FDASIA
- 2016: 21st Century Cures

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.