Annotating the literature on pathogenicity of PRNP variants
In our study last year, by comparing case and population control allele frequencies, we managed to come to conclusions about the penetrance of only 14 out of the now 67 reportedly pathogenic PRNP variants [Minikel 2016]. The remainder were too rare, both in prion disease cases and population controls, for allele frequency comparisons to reach any meaningful conclusion. But it would be very useful to have at least an educated guess about the penetrance of the other 53 variants. Some are probably high penetrance (say, >50% lifetime risk), others might confer a risk that is increased above the population baseline but still low, and some might be completely benign. Where to begin?
To start to get at this problem, I recently took a deeper dive on the literature about these 67 reportedly pathogenic variants, looking at the human genetic evidence for each variant’s pathogenicity. Here are the criteria I looked at:
- Mendelian segregation. In a group of closely related individuals, if most or all of the individuals with a particular genetic variant develop a particular disease, and the people without that genetic variant do not develop the disease, this is called Mendelian segregation. Every geneticist agrees that segregation is a valuable clue in investigating pathogenicity, but the context matters a lot. The American College of Medical Genetics (ACMG), for instance, considers segregation to be only “supportive evidence” (as opposed to “strong” or “very strong” evidence) of pathogenicity, because all it means is the locus (a segment of a chromosome) is linked to disease, it doesn’t prove that one specific variant is causal [Richards 2015]. But in prion disease, there is only one causal gene, PRNP, all known pathogenic variants therein are protein-coding, and there is just one short open-reading frame that can easily be sequenced. The upshot is that if a rare protein-altering variant in PRNP segregates with prion disease, it is the causal variant. Investigators looking to establish a novel gene as disease-causing might like to see segregation in two different families before they’re confident, but for a novel variant in a well-established disease gene, I think that segregation even in one family (especially if that’s the only family with the variant) is pretty strong evidence (perhaps not definitive proof) of fairly high penetrance. I therefore looked through the literature to see, for each variant, if there was even one family where there were at least three closely related affected individuals in a pattern consistent with Mendelian segregation. If there was, I considered this evidence for high penetrance.
- De novo variants. Just because a disease is genetic doesn’t mean it’s inherited — the average person has ~60-70 de novo (spontaneous) mutations in their genome, mutations that neither of their parents had, ~1 of which falls in a protein-coding portion of a gene [Michaelson 2012, Kong 2012]. And if a person with a de novo mutation in PRNP has prion disease, that mutation is probably highly penetrant. As a back of the envelope, if the average person has only about 1 protein-coding de novo SNP or indel in their entire genome, and there are ~20,000 genes, of which PRNP is one of the smaller ones, and only ~20,000 prion disease cases have ever come to the attention of the modern medical establishment. Therefore, it’s unlikely there’s ever even been one individual who had sporadic prion disease and just happened to have a benign de novo in PRNP just by coincidence. Others who do variant classification seem to agree — ACMG considers de novo status to be “strong evidence” of pathogenicity [Richards 2015]. I considered de novo status to be evidence for high penetrance.
- Homozygotes. Almost all cases of genetic prion disease are in heterozygotes — people with just one mutant copy of the PRNP gene. But a few variants have been seen in homozygotes. For E200K, which has ample evidence for high penetrance and is found in some dense founder clusters around the world [Lee 1999], this isn’t too surprising. But for a couple of variants that don’t have evidence for high penetrance, the presence of an affected homozygote is suggestive that the variant at least confers an increased risk of prion disease. That’s because these PRNP variants are so rare that even one affected homozygote can represent a very unlikely-by-chance deviation from Hardy-Weinberg equilibrium. Let me explain. The variants in question have allele frequencies «0.1% in the general population (based on ExAC continental populations). A variant with, say, AF 0.1% has a het frequency of 2 in 1,000 and, under random breeding, a homozygote frequency of 1 in 1,000,000, so there are 2,000 hets out there for every 1 homozygote. Consider that 1 of the 4 known affected V203I individuals is a homozygote [Komatsu 2014], and 1 of 3 affected Q212P individuals is a homozygote [Beck 2010, Minikel 2016]. In both cases, the homozygotes had no family history of the disease on either side of the family. Without doing any math, it’s clear that these numbers are fairly unlikely to happen by chance if the variant confers no risk. Here, a likely explanation is that one mutant allele confers an elevated but still low risk, while two mutant alleles confer a higher risk. Thus, in these cases, an affected homozygote provides some evidence that a variant confers risk increased above the baseline.
- Case/control enrichment. For completeness, I also noted the handful of variants for which we have evidence that the variant is more common in cases than controls [Minikel 2016], as this is evidence for increased risk or, with very strong enrichment, high penetrance.
All that said, here’s what I found:
This table was last updated 2025-01-21. If you use these data please cite the latest published version: [Goldman & Vallabh 2022].
| variant | evidence for high penetrance | evidence for increased risk | refs | comments |
|---|---|---|---|---|
| P39L | Bernardi 2014 | |||
| 2-OPRD | Beck 2001, Capellari 2002 | |||
| 1-OPRI | Laplanche 1995, Pietrini 2003 | |||
| 2-OPRI | Hill 2006 | |||
| 3-OPRI | Nishida 2004 | |||
| 4-OPRI | Kaski 2011 | most cases have negative family history | ||
| 5-OPRI | Mendelian segregation | Mead 2007 | ||
| 6-OPRI | Mendelian segregation | Mead 2006 | ||
| 7-OPRI | Mendelian segregation | Goldfarb 1991 | ||
| 8-OPRI | Mendelian segregation | Goldfarb 1991, Laplanche 1999 | ||
| 9-OPRI | Mendelian segregation, de novo | Krasemann 1995, Sanchez-Valle 2008 | ||
| 12-OPRI | Mendelian segregation | Kumar 2011 | ||
| P84S | Jones 2014 | |||
| S97N | Zheng 2008 | |||
| P102L | Mendelian segregation | case/control enrichment | Webb 2008 | |
| P105L | Mendelian segregation | Yamada 1999 | 2 sibs affected & genotyped, 1 ungenotyped parent likely affected | |
| P105S | Tunnell 2008 | |||
| P105T | Mendelian segregation | Rogaeva 2006 | ||
| T107I | Mendelian segregation | Holm-Mercer 2025 | ||
| G114V | Mendelian segregation | Rodriguez 2005, Liu 2010 | pedigree suggests penetrance high though not 100% | |
| A117V | Mendelian segregation | case/control enrichment | Hsiao 1991 | |
| 129insLGGLGGYV | de novo | Hinnell 2011 | ||
| G131V | Panegyres 2001, Jansen 2011 | positive family history in one case | ||
| G131R | Alshaikh 2020 | positive family history | ||
| S132I | Mendelian segregation | Hilton 2009 | extensive family history, only proband genotyped | |
| A133V | Rowe 2007 | |||
| R136S | 2 homozygotes | Ximelis & Moreno 2021 | ||
| Y145X | Kitamoto 1993 | |||
| R148H | Krebs 2005 | |||
| R156C | Kenny 2017 | |||
| Q160X | Mendelian segregation | Fong & Rojas 2016 | ||
| Y162X | Mendelian segregation | Bommarito 2018 | ||
| Y163X | Mendelian segregation | Mead 2013, Capellari 2018 | ||
| D167G | Bishop 2009 | |||
| D167N | Beck 2010 | |||
| Y169X | Mendelian segregation | Capellari 2018 | ||
| V176G | Simpson 2013 | |||
| D178Efs25X | Mendelian segregation | Mastuzono 2013 | only proband genotyped | |
| D178N | Mendelian segregation, de novo | case/control enrichment | Medori 1992, Dagvadorj 2002 | |
| V180I | case/control enrichment | Hitoshi 1993 | ||
| T183A | Mendelian segregation | Nitrini 1997 | ||
| H187R | Mendelian segregation | Butefisch 2000 | ||
| T188A | Collins 2000 | |||
| T188K | Roeber 2008 | multiple cases with negative family history | ||
| T188R | Roeber 2008, Tartaglia 2010 | |||
| V189I | Di Fede 2019 | |||
| T193I | Kotta 2006 | |||
| K194E | Takada 2017 | |||
| E196A | Zhang 2014 | |||
| E196K | Mendelian segregation | Peoc’h 2000 | only proband genotyped | |
| F198S | Mendelian segregation | Dlouhy 1992, Hsiao 1992 | ||
| F198V | Zheng 2008 | |||
| E200D | homozygote | Hassan 2021 | ||
| E200G | Kim 2013 | |||
| E200K | Mendelian segregation | homozygote, case/control enrichment | Hsiao 1991 | |
| T201S | Parvez 2010 | |||
| D202G | Mendelian segregation | Heinemann 2008 | only proband genotyped | |
| D202N | Piccardo 1998 | |||
| V203I | homozygote | Komatsu 2014 | ||
| R208C | Zheng 2008 | |||
| R208H | Mastrianni 1996 | |||
| V210I | case/control enrichment | Ripoll 1993, Pocchiari 1993 | ||
| E211D | Mendelian segregation | Peoc’h 2012 | supplement describes 1 family with 3 affected | |
| E211Q | Peoc’h 2000 | 2 sibs affected | ||
| Q212P | homozygote | Beck 2010 | ||
| I215V | Munoz-Nieto 2013 | |||
| Q217R | Hsiao 1992 | 2 affected | ||
| Y218N | Mendelian segregation | Alzualde 2010 | ||
| A224V | Watts 2015 | |||
| Y225C | Bagyinszky & Yang 2019 | |||
| Y226X | Jansen 2010 | |||
| Q227X | Jansen 2010 | |||
| M232R | case/control enrichment | Hitoshi 1993 | ||
| M232T | Bratosiewicz 2000 | |||
| P238S | Windl 1999 |
In total, then, 27 out of the 74 have evidence for either Mendelian segregation or de novo status according to these criteria. These are all likely to be high penetrance variants. For some of these we can say definitively that penetrance is high, when the family is large or when there is dramatic case/control enrichment. For the rest it’s likely, although it’s conceivable for some variants that the penetrance is somewhat more modest and maybe there just happened to be a family with three affecteds by coincidence.
There are also probably some variants that are genuinely high penetrance but have no boxes checked above. For instance, sometimes a family history just isn’t available for a patient, or there is a history of disease but the family never speaks about it and so the younger generation doesn’t know, or the family history appears negative only due to adoption or a non-paternity event, or the variant is de novo but the parents are already deceased and there are no siblings, so it is impossible to prove that it’s de novo. When there’s only one patient and the variant is also ultra-rare in controls, it’s hard to say anything completely definitive.
Overall, then, I am not asserting that any of the criteria above are proof positive for a particular risk classification, but I think they each make a classification more or less likely, and are worth noting. Many papers on genetic prion disease include a figure with a diagram of PRNP’s coding sequence, sometimes with elements of protein secondary structure noted, and all the reportedly pathogenic mutations indicated. I set out to make a new such figure with variants shaded by their level of human genetic evidence (evidence for high penetrance, evidence for increased risk, or no evidence) and sized by the number of cases in our recent case series [Minikel 2016]. Here’s what I came up with:
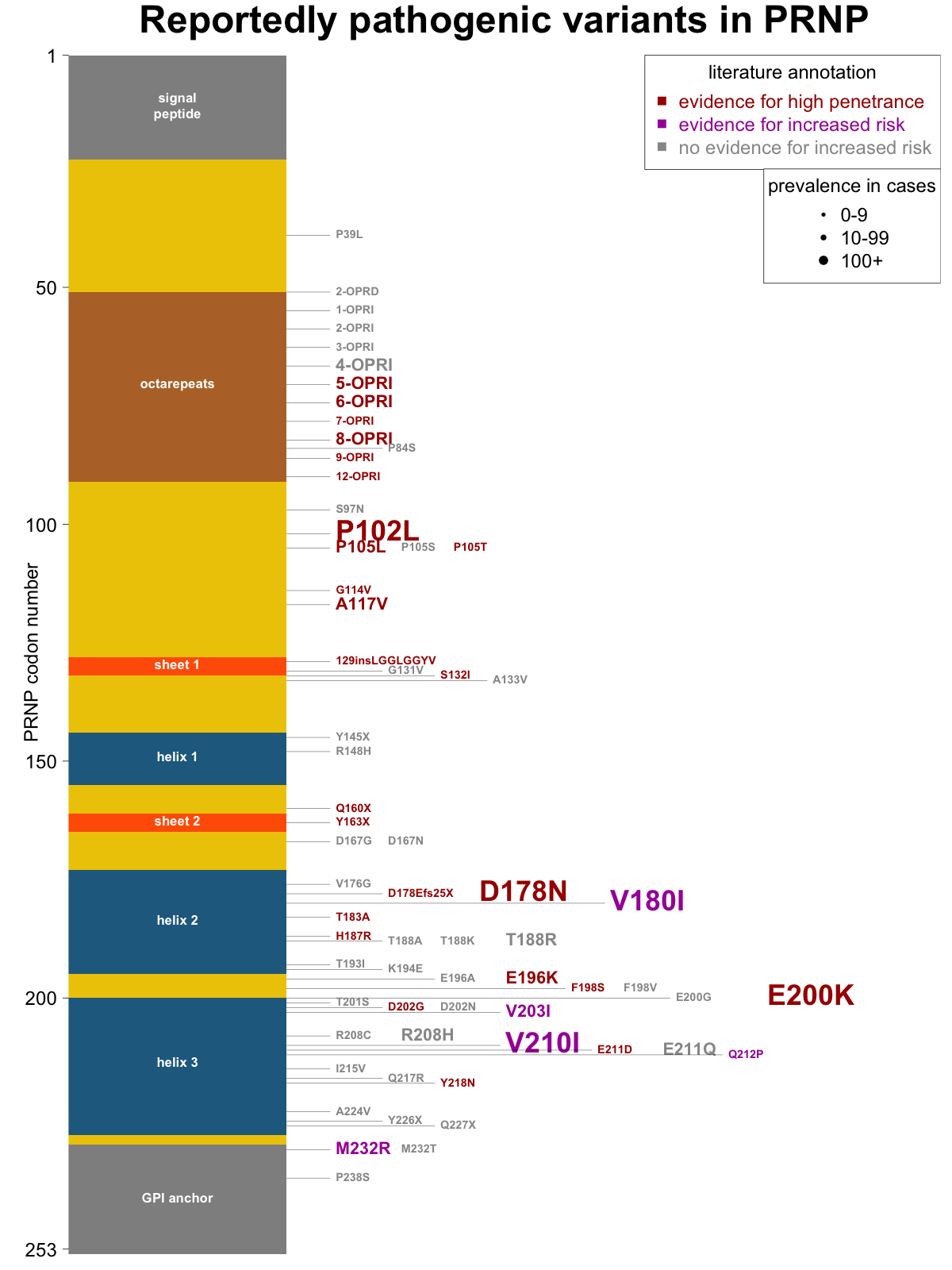
As a future direction, there are additional sources of information that should be useful in classification that I haven’t gotten into here but may go through and annotate in the future:
- More could probably be done with allele frequency comparisons — for instance in some cases the sheer frequency in controls is too high for a variant to be highly penetrant, and can be enough to suggest that certain variants are likely benign.
- Multiple isolated cases without family history provide some evidence against high penetrance.
- CpG variants with a low case count are probably not high penetrance. CpG variants (C → T transitions where the next base is G) are the most frequent type of DNA mutation, occurring 10X more often than non-CpG transitions and 100X more often than transversions [Samocha 2014, Lek 2016]. CpG variants are responsible for all three of the most prevalent, most highly recurrent PRNP mutations in cases (P102L, D178N, and E200K). Based on mutation rates, we can estimate that other CpG variants in PRNP have probably arisen very roughly about as many times in the world population as those three, so they’ve had roughly as many chances to produce prion disease cases, if they were highly penetrant. So if you see a CpG variant that has few cases, and no Mendelian segregation, it’s likely to confer at worst a low risk of disease, and perhaps no risk at all.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.