Human prion propagation in cell culture
It’s time to talk again about cell culture systems as tools for prion disease research. I blogged a “brief history” of this topic four years ago, which remains a decent read on the subject, but I missed a few interesting lines of research, and some interesting new developments have happened since 2013.

Above: some human IMR32 neuroblastoma cells that are, like all human immortalized cell lines, not propagating prions.
For starters, let’s recap that brief history in even briefer form. For 30 years now, we’ve had ScN2a cells — mouse neuroblastoma cell lines infected with mouse RML prions [Race 1987, Race 1988, Butler 1988], and those cell lines have proven to be really useful tools, but with key limitations. Phenotypic screening in these cells has, on multiple occasions, allowed the discovery of antiprion small molecules that then proved effective against mouse prions in vivo, extending the survival time of infected mice by 2-4X [Kawasaki 2007, Wagner 2013, Berry 2013, Giles 2016]. But none of those compounds work against human prions in humanized mice [Berry 2013, Lu & Giles 2013, Giles 2015, Giles 2016]. This problem leads to the thought: if only we had the ability to do phenotypic screening in human cells infected with human prions, maybe we’d have a small molecule in clinical trials by now.
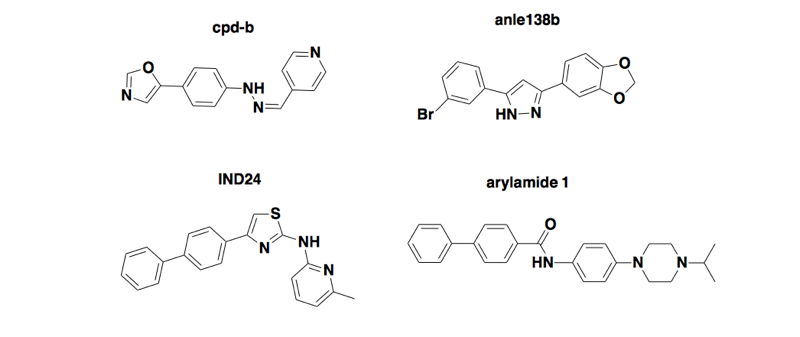
Above: four compounds discovered by phenotypic screening in mouse cells infected with mouse prions, that extend survival in wild-type mice infected with mouse prions but don’t work in humanized mice infected with human prions [Berry 2013, Lu & Giles 2013, Giles 2015, Giles 2016].
So why don’t we have the human analog of those ScN2a cells? It’s not for lack of trying. Though next to nothing has been published on the subject, my informal survey of my colleagues in the prion field suggests that a number of people have tried the same approach that gave rise to the ScN2a cells. Which is: dump prion-infected brain homogenate on cells expressing human PrP, passage them enough times to dilute the original prion material to a negligible quantity, possibly subclone to try to identify and expand individual clones that are infected, and Western blot after proteinase K digestion to see if you’ve got new prions generated by the cells.
And it’s also not that there’s something unique about mouse prions that makes infection possible. If anything, the data would hint that there’s something unique about human prions that makes infection impossible, because at this point people have succeeded with virtually every oft-studied species except human. For instance, prions have been propagated in cell lines from hamster [Taraboulos 1990], rat [Rubenstein 1992], deer [Raymond 2006], and elk [Bian 2010]. The cell line RK13, which is derived from rabbit kidney epithelial cells and has little or no endogenous PrP expression, has proved to be an especially versatile tool. Transfect them to express sheep PrP they can be infected with sheep scrapie prions [Vilette 2001], express elk PrP and you can propagate elk CWD prions [Bian 2010, Kim 2012], express goat PrP and you can propagate goat prions [Dassanayake 2016]. Express bank vole PrP, which is an apparently universal acceptor for prions in vivo [Watts 2014], and you can infect with a variety of bank vole-adapted prion strains, including cattle and sheep strains [Courageot 2008]. But express human PrP, and still no propagation of human prions, even though the same human prions, passaged through mice and then dumped onto RK13 cells expressing mouse PrP, do propagate [Lawson 2008]. It’s immensely frustrating.
When I sat down a couple of years ago to ponder why this is, and what might be done to solve it, I came up with the following hypotheses about what was going wrong, and how to fix it.
First, there is a potential issue with the kinetics of prion replication. Prions are rarely transmitted horizontally between cells in culture, but rather are passed down through cell division, so infection can only be maintained if the rate of prion formation exceeds the cumulative rate of prion clearance and cell division [Ghaemmaghami 2007]. Anecdotally, I’ve heard that even ScN2a cells can be finnicky, sometimes losing infection after freeze-thaw or if they are passaged too rapidly, suggesting that even in ScN2a cells, the kinetics only just barely work out. And human prions replicate more slowly than mouse prions — I base this assertion on the fact that HuPrP knock-in mice, which express human PrP at normal levels, take ~500 days to develop prion disease for most human prion strains (though MV2 and VV2 are notable exceptions at just under 300 days) [Bishop 2010, see Table 1], whereas wild-type mice, expressing mouse PrP at normal levels, develop symptoms of RML prion infection in about 130 days [Fischer 1996]. Similarly, the highest-overexpressing human PrP mice still take at least 160 days to develop prion disease after inoculation with human prions [Telling 1995, Kong 2005, Beringue 2008, Race 2009], while the highest-overexpressing mouse PrP mice, such as Tga20 and Tg4053, get sick from RML prions within just 50 - 60 days [Fischer 1996, Telling 1996]. So: maybe human prions simply can’t replicate fast enough to keep up with cell division in immortalized cell lines under usual culture conditions. If so, maybe keeping the cells stationary for periods of time or passaging less aggressively would help. Or maybe some prion infectivity is being propagated, but at low levels, and so a more sensitive detection method is needed.
Second, maybe human cells primarily propagate protease-sensitive prions. In the brains of people who died of sporadic CJD, about 90% of the PrPSc is protease-sensitive [Safar 2005], and there are other subtypes of prion disease in which there is little or no protease-resistant PrP [Medori 1992, Brown 1995, Zou 2010]. Prions have sometimes been seen to lose their protease resistance, for instance, upon passage into a new species or PrP sequence background [Lasmezas 1997, Leske 2017], and indeed, the earliest prion-infected N2a cells were determined to be propagating infectivity by bioassay but did not exhibit protease resistance [Race 1987]. So what if human cells are propagating prions, but we’re missing it because we’re only looking by PK digestion and Western blotting? Again, I reasoned that maybe a new detection method is needed.
Based on the above lines of reasoning, I convinced myself that maybe the key was to start looking for prion infection in cultured cells using RT-QuIC, which detects protease-sensitive prions [Vascellari 2012] and is far more sensitive than Western blots, because it relies on exponential amplification [Wilham 2010].
But it turns out that the people who have now started to have some success with propagating human prions in culture have had a totally different approach: backing away from immortalized cell lines and focusing on primary cultures or stem cell-derived cultures, in a bid to get as close as possible to the authentic cell types susceptible to prion infection in vivo.
This line of thinking may go back even farther, but from what I can tell, one of the earliest hints that this approach would bear promise came from a study of sheep prions [Cronier 2004]. They created primary cultures from the cerebella of transgenic mice expressing sheep PrP and infected them with sheep prions. Not only did they see prion propagation, they saw neuronal death, which is not observed in ScN2a or other immortalized cell lines infected with prions. A few years later, the same group reported that they had tried the same trick with primary cultures of cerebellar granule neurons (CGNs) from mice overexpressing mouse, hamster, or human PrP, and had succeeded in each case [Cronier 2007]. They didn’t demonstrate infectivity by bioassay or re-infection of cells using infected cell lysate, and there wasn’t a deep characterization of the kinetics or the sustainability of infection in these cultures over time. But they showed Western blots that Tg(HuPrP) CGN cultures exposed to CJD brain homogenate had very low PK-resistant PrP after 14 days, and much higher levels after 28 days, which certainly suggests that prion replication was occurring. And another group later replicated their findings using a very similar system with CGNs from two different transgenic HuPrP mouse models, and four different human prion isolates, and with a bit more characterization of the kinetics too [Hannaoui 2014].
And it makes sense that that approach might work, as there have been other success stories with modeling prion disease in primary culture as well. The best known system is the cerebellar organotypic cultured slice (COCS) system, where whole 350 μm cross-sections of neonatal mouse cerebellum are maintained in culture for weeks [Falsig & Aguzzi 2008]. It’s practically a whole brain in culture — you have multiple cell types and different layers and so on — and it was first shown to replicate prions [Falsig 2008] and later to recapitulate neuronal death as well, if the slices could be kept alive in culture long enough [Falsig 2012].

Above: neuronal damage (white) in the cerebellar granule cell layer of a cerebellar organotypic cultured slice infected with RML prions. From Figure 1 of [Falsig 2012].
So if these cerebellar slices can model prion disease in culture, is the full brain tissue context with different cell types essential, or could it be pared down at all? Could a monoculture of one cell type do it? Do the cells need to be post-mitotic? There has been success at propagating mouse prions in mouse neurospheres (clusters of partially, but not terminally, differentiated neural precursor cells) [Giri 2006] and in “brain aggregates” containing neurons and glia [Bajsarowicz 2012, Tousseyn 2015]. Some level of cell differentiation is probably key to these successes. One study found that embryonic stem cells express only low levels of PrP, and that expression goes up several-fold after directed differentiation [Krejciova 2011]. A careful time series study of human embryonic stem cells showed that they actually do take up prions after exposure to infected brain homogenate, but they clear the prions within about 72 hours [Krejciova 2011].
Which brings us to the latest chapter in this saga: successful human prion propagation in cultured astrocytes derived from induced pluripotent stem cells. I blogged about this work after Zuzana Krejciova presented it at Prion2016 in Tokyo and it was published last month [Krejciova & Alibhai 2017]. They worked with human induced pluripotent stem (iPS) cell cultures and used established protocols to differentiate them first into dividing, partially differentiated astrocyte progenitor cells (APCs) and then into mostly non-dividing, terminally differentiated astrocytes. When the cells were exposed to brain homogenate from human sporadic or variant CJD brains, the APCs failed to propagate (or even really take up) any prions, but the terminally differentiated astrocyte cultures showed a clear increase in PK-resistant PrP from day 0 to day 3 to day 8 all the way up to day 28 post-exposure. And, although it’s an N of just 1 or 2 for each genotype, the infection did appear to be genotype-dependent in exactly the way you’d expect: vCJD, which has affected almost exclusively PRNP 129MM individuals [Mok 2017], readily propagated only in a line of 129MM astrocytes. It propagated more slowly and less reliably in 129MV and not at all in 129VV astrocytes. Prions from an sCJD VV2 case propagated relatively quickly in 129VV astrocytes but much more slowly in 129MM astrocytes. They also used lysate from infected cells to re-infect other cells, in order to show that there was really infectivity and not just protease resistance.

Above: 3D reconstruction of PrPSc deposits (green) on cultured human astrocytes (nuclei stained blue). Courtesy of Zuzana Krejciova.
It’s intriguing and surprising that all this worked in astrocytes, which I don’t normally think of as the main cell type involved in prion disease. The accompanying commentary on this study [Aguzzi & Liu 2017] goes into detail on this issue and has helped to resolve a cognitive dissonance I’d been holding in my head for years.
As background, neurons are the only cell type that degenerates or dies as a result of prion infection [Liberski 2004], although there are plenty of other cell types that can support prion infection, such as B cells [Klein 1997] and follicular dendritic cells [Brown 1999, Montrasio 2000]. It appears that prion toxicity is cell autonomous [Brandner 1996], so it stands to reason that if you want to prevent or treat prion disease by lowering or eliminating PrP, it’s the neurons that you need to reach. And indeed, when PrP was conditionally deleted using the Cre system under the NFH promoter, which is active only in neurons, mice were saved from dying of prion disease, even while they continued to accumulate PrPSc in their brains, presumably because glia were still replicating prions [Mallucci 2003]. Yet, in direct contradiction with those findings, other studies had found that expression of PrP under the supposedly astrocyte-specific GFAP promoter was sufficient to render mice susceptible to infection with prions and death from prion disease [Raeber 1997, Kercher 2004, Jeffrey 2004]. So which answer is correct — is astrocyte-specific expression of PrP enough to cause susceptibility to prion disease? The commentary [Aguzzi & Liu 2017] resolves this dilemma, by pointing out that the GFAP promoter used in those studies was later found to express in many neurons, and not only in astrocytes as originally assumed [Zhuo 2001, Casper & McCarthy 2006]. Thus, the original assumption still stands — the evidence indicates that PrP expression in neurons is required for prion neurotoxicity. But, astrocytes do replicate prions (apparently unlike microglia), and the new study [Krejciova & Alibhai 2017] provides further evidence of this.
Where does the recent progress put us in terms of drug discovery? The notion of repeating, this time with human cells, the phenotypic screens that led to the discovery of antiprion compounds effective in mice, might finally be inching within reach. To be sure, working with iPS cells and differentiation protocols is a ton of work. I’ve never actually been responsible for them myself, but I’ve seen labmates grow iPS cells, and the cells demand hours of care each day. Just following the differentiation protocols to be able to start your experiment can take weeks. The astrocyte differentiation protocol used in the new study, for example, takes 2 weeks [Serio 2013]. But some of the antiprion compounds discussed at the top of this post were actually surprisingly low-hanging fruit. IND24, for example, which can quadruple survival time in prion-infected mice if given prophylactically [Giles 2015], came out of an academic screen of just 10,000 compounds [Ghaemmaghami 2010]. That’s tiny compared to the screens routinely done at pharmaceutical companies. I corresponded with Zuzana Krejciova, lead author of the new study, and she said she has had in culture as many as 100x 10cm plates of astrocytes at once, and that, with a lot of effort, a 10,000-compound screen in this model might be feasible.
There would certainly be kinks to work out — getting good results out of a screen means you need a good Z factor [Zhang 1999], which basically boils down to, you want the negative controls (cells without compound) to be virtually identical to each other, so that if a compound does anything to the cells, it stands clearly out from the crowd. When you’re working with relatively high-maintenance cells like iPS-derived astrocytes, there’s a risk that each group of cells turns out a little different, and it can be difficult to develop a screen with good statistical power. Still, whether the new cell model does end up being scaled for primary screening, or ends up as a tool for testing tens or hundreds of compounds in a secondary screen, I hope that it will prove useful, and that over the coming years we will start to see more examples of candidate antiprion compounds being tested in human cell culture.
Zuzana Krejciova provided feedback on a draft of this post.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.