Is PRNP associated to any human traits besides prion disease?
introduction
I’ve now been studying the prion protein (PrP) and its human gene (PRNP) for nearly 8 years, and to the best of my knowledge, prion disease is the only human phenotype that has ever been convincingly associated to PRNP. But with genome-wide association studies (GWAS) getting ever larger and better-powered, and with some scientists speculating that many complex traits will turn out to be virtually “omnigenic” [Boyle 2017], it is good to check one’s assumptions from time to time. In this post, I’ll set out to assess the evidence for whether PRNP is associated to any phenotype other than prion disease, and in the process, I will explore a lot of what remains so challenging about mapping GWAS association signals to causal genes.
PRNP associations in the GWAS Catalog and Open Targets Genetics
I first looked in the EBI GWAS Catalog, which aggregates GWAS hits from published literature. A quick search for PRNP did not reveal anything unexpected. The catalog includes four GWAS studies associating PRNP to, as expected, the risk of sporadic and variant Creutzfeldt-Jakob disease (two subtypes of prion disease) [Mead 2009, Mead 2012, Sanchez-Juan 2012, Sanchez-Juan 2015]. There is also one study associating PRNP to the KANNO blood group [Omae 2019]. KANNO is an interesting story and one that I believe to be true (I will explain more in an upcoming post), but it’s not actually a disease or even a trait per se, just a molecular measurement of PrP antigenicity in blood. Thus, the GWAS Catalog did not suggest any new or unexpected phenotypic associations to PRNP.
But the GWAS Catalog is limited in a few ways. For one, it only includes studies published in the form of peer-reviewed journal papers. The massive UK BioBank GWAS undertaken by the Neale Lab and the Lee Lab are not captured. Equally important, the GWAS Catalog incorporates only the most straightforward assignment of hit SNPs to causal genes. In the GWAS Catalog, a gene may be denoted for a SNP if the authors of the published study report that gene as being causal (based on whatever data or criteria they deem fit), or, failing that, if it is closer to that gene than to other genes. This could mean that some SNPs that affect a phenotype through their effects on PRNP but are non-coding and relatively distant from the PRNP transcription start site might not be assigned as belonging to PRNP.
I therefore also took a look at PRNP on Open Targets Genetics (OTG), a collaboration undertaking an enormous and highly sophisticated GWAS data integration effort, serving up the UK BioBank GWAS results in a user-friendly browser while incorporating the latest and greatest in causal gene assignment for GWAS hits. I was surprised to find that, on the PRNP page under Associated Studies, OTG lists no fewer than 59 hits, spanning a wide variety of phenotypes. 4 are hits for Creutzfeldt-Jakob disease, as expected, but the remaining 55 are dominated by hematologic phenotypes such as reticulocyte count, immature reticulocyte fraction, high light scatter reticulocyte count, and mean corpuscular hemoglobin. The sources of these hits included UK BioBank as well as published literature. These associations seemed a priori unlikely to me, because PrP has very low expression in human blood [Vallabh 2019], and because associations of PrP to hematologic phenotypes in animal models have so far not been convincing [Wulf 2017] (more on that later in this post). Moreover, whereas the lead SNPs for prion disease are located in or near the PRNP coding sequence, many of the lead SNPs for hematologic phenotypes were 500 kb away, just upstream of another protein-coding gene, SMOX. While these considerations do not rule out a genuine association to PRNP, they motivated me to investigate further.
The more I learned, the more I realized that the assignment of SNPs to causal genes is a truly thorny problem that I should take the time to learn a bit more about.
assigning GWAS hits to causal genes
Why is it so hard to determine which gene is responsible for a GWAS hit?
In Mendelian disease, most causal variants are either protein-coding or have other obvious links to a single gene. They are rare, their effect sizes are large, and Mendelian segregation in just a few families can convincingly implicate a gene. GWAS is different. Most hits are common variants of small effect size. Each “hit” is not a single SNP but, rather, a linkage disequilibrium (LD) peak — a whole collection of SNPs that have traveled together with the (often single) true causal SNP on a haplotype block across human generations. And most genome-wide significant SNPs are non-coding, and these variants can sometimes have regulatory effects at a considerable distance.
This complicates gene assignment in at least two ways. First, an LD peak may be physically large — the set of genome-wide significant SNPs for one peak can sometimes span several genes. Second, the potential for non-coding SNPs to have regulatory effects at a distance means that even if a true causal SNP can be identified, the nearest gene, while certainly a decent first guess, does not always turn out to be the gene through which that SNP acts.
If you really, really want to know which SNP, and which gene, are responsible for a GWAS hit, you can easily spend a whole postdoc — or more — on finding out the answer, through detailed functional studies in cell culture and/or animal models.
One example is a study that assigned an LD peak for type 2 diabetes (T2D) on chromosome 17 to SLC16A11 [Rusu 2017]. When this peak was first identified as a GWAS hit [Hara 2014], the only genome-wide significant SNP was in the gene SLC16A13, though the apparent peak was broad and included suggestive P values spreading into neighboring genes. With larger sample sizes plus exome sequencing and imputation to identify additional SNPs, it became possible to fine-map the hit to a “99% credible set” — a shortlist of SNPs expected to be very likely to contain the true causal variant. All of the variants that made the shortlist turned out to be in or near SLC16A11, not SLC16A13. There followed years of painstaking analysis of RNA expression levels, epigenetic marks, and characterization of protein behavior after expression of different variants in cell culture models. The authors determined that the T2D risk haplotype affected the plasma membrane localization of the SLC16A11 protein by disrupting an interaction with a binding partner, and that this change in localization in turn affected lipid metabolism and T2D risk.
Another example is the study that implicated an LD peak for obesity on chromosome 16 as affecting brown fat through regulatory action on IRX3 and IRX5 [Claussnitzer 2015]. The lead SNP for that peak was physically located in a gene called FTO, and in fact, FTO got its name (“fat mass and obesity-associated protein”) from the presumption that it was causal. But through a combination of CRISPR editing in human cell lines, knockdown and overexpression experiments, and mouse models the authors showed that the causal variant disrupted a transcriptional repressor, thus affecting expression of IRX3 and IRX5, two genes located a whopping 1-2 Mb away, that appear to provide master control over a “browning” program in fat cells that affects the rate at which we burn energy.
Those are both fascinating studies, and I would argue that we as a scientific community should do more to support and reward studies like these. Really dissecting mechanism like this is the way that we will get to credible therapeutic hypotheses, and I sometimes worry that deep (thorough dissection of a single hit or single gene) science gets too little credit relative to the wide (genome-scale) science.
Regardless, those types of truly deep studies are only ever going to be done on a short list of very interesting GWAS hits, not on every one of the thousands and thousands of hits that GWAS are churning out these days. So for those operating at a genome-wide scale, or for those of us who don’t have five years and a million dollars to spend, how can we get at least a fairly educated guess as to what gene is causal for a given hit?
There are a few solutions out there.
One is fine-mapping — as sample sizes get larger, and provided that sequencing or imputation can more exhaustively identify all of the candidate SNPs on the haplotype, rare recombination events will pile up, helping to make the causal SNP stand out above the passenger SNPs that usually travel on its haplotype [Huang 2017]. Narrowing a hit down to a single very likely causal SNP can in some cases (though not always) convincingly implicate one gene.
Another solution is functional genomics — you ask whether the GWAS hit SNPs are also known, from other datasets, to have functional effects on a gene. See, as one example, a recent study of drug targets for immune-related traits [Fang 2019]. Potential sources of functional genomics data include expression quantitative trait locus (eQTL) data from GTEx, proximity of SNPs to gene promoters in 3-D space using Hi-C, and presence of SNPs in enhancer regions based on ENCODE data, among others. So, for instance, if your lead SNP associated to your favorite trait (YFT) is also associated to expression of your favorite gene (YFG) in a functional genomics dataset (and especially if that association is present in a disease-relevant tissue), then you might be relatively more confident that YFG is the gene that controls YFT.
But mere overlap between GWAS hit SNPs and functional genomics hit SNPs is not necessarily enough to prove that a GWAS hit can be assigned to a given gene. Consider the below example:
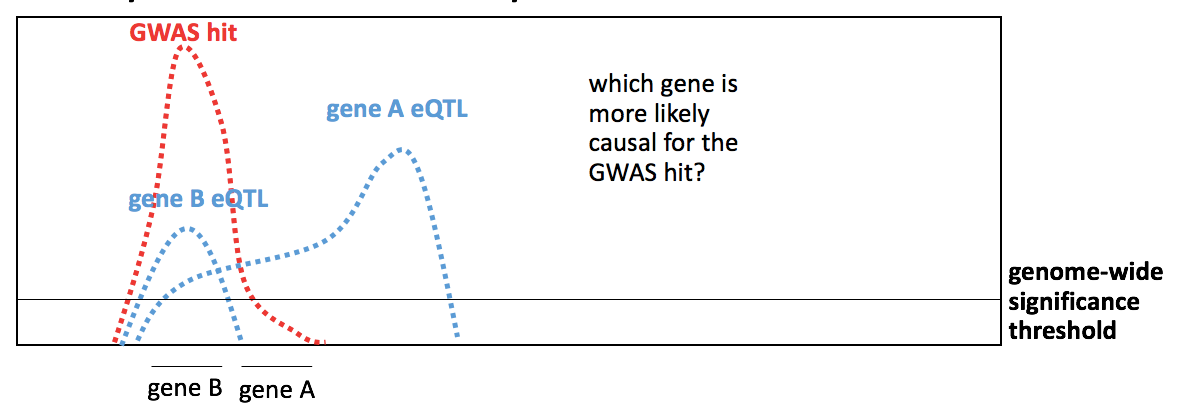
The GWAS hit LD peak overlaps with the eQTL LD peak for gene A, and indeed, some of the exact same SNPs may be genome-wide significant for both the GWAS and the gene A eQTL. So if your analysis is based on overlap alone — intersecting the list of genome-wide significant SNPs — you will conclude that gene A is the, or a, causal gene. But the shape of the LD peak for the gene A eQTL is actually very different from the shape of the GWAS LD peak — it looks like the true causal SNP for the gene A eQTL is likely to be further to the right, while the intersection with the GWAS hit is just a coincidental overlap at the peak’s shoulder. Instead, the GWAS hit looks like it is almost the same shape as the eQTL hit for gene B, making B the more likely causal gene. This intuition is formalized in a concept called colocalization [Giambartolomei 2014], a statistical method for determining whether the shape and overlap of two LD peaks is sufficient to make them likely to be driven by the same underlying causal variant. Performing a full colocalization analysis requires access to full summary statistics for both association studies, which is computationally a heavy lift, and often impossible because (very unfortunately) not all GWAS authors release their summary statistics. There are workarounds, though — a recent method called PICCOLO [Guo & Sieber 2019] appears to be able to achieve 89% specificity and 52% sensitivity using just the GWAS index SNP, by leveraging known LD patterns to infer what the LD peak for that hit might look like.
With these concepts in hand, I went back to explore in more detail the PRNP associations in the Open Targets Genetics portal.
Analysis of PRNP data in Open Targets Genetics
I started by examining the OTG Associated Studies table:
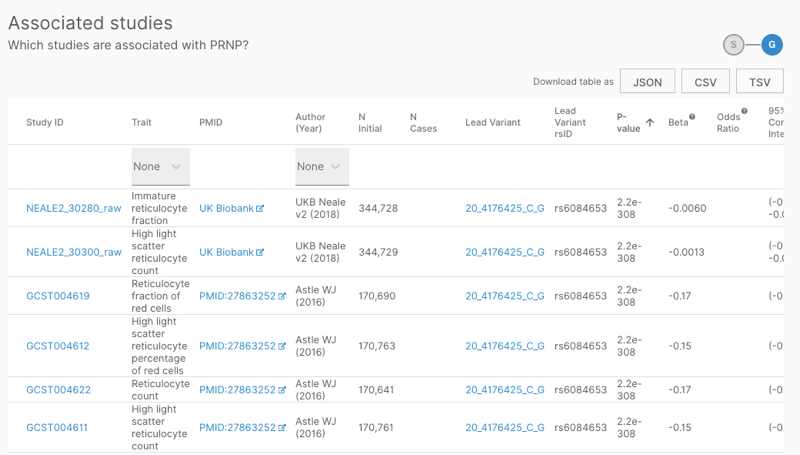
This table provides no information on what type of evidence, or what strength of evidence, relates the hit SNPs to the gene of interest. A look at one of the published GWAS studies cited [Astle 2016] revealed that it reported only the SNPs and made no speculation as to causal genes. The Ben Neale UK BioBank GWAS, a source of many of these associations, also does not assign causal genes. The table can be downloaded as a TSV file, which contains additional columns, but none of these provided more explanation as to the gene assignments.
Clicking on the “Lead variant” link in that table leads to a variant page, such as that for 20_4176425_C_G, which is the lead SNP for the top GWAS hit, immature reticulocyte fraction in UK BioBank. At the top of this page is an “Assigned genes” table summarizing OTG’s variant-to-gene (v2g) inferences:
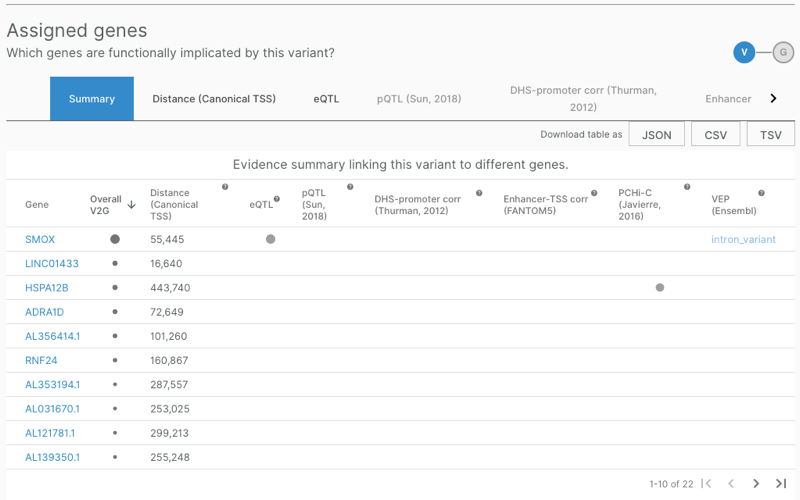
These v2g inferences are based on the degree to which functional genomics data implies that a given variant is related to a given gene — unlike true colocalization analysis, the inference is not specific to a particular GWAS study in which the variant was identified as a hit. They show data for individual sources of functional evidence (GTEx, VEP, Hi-C, and so on) as well as a total score, overall_v2g. A glance at the overall scores for 20_4176425_C_G, visualized as dots of varying sizes, reveals a high score for SMOX and rather low scores for the next 9 genes. Clicking through the additional pages of results reveals that PRNP is not even among the 22 genes listed for this SNP.
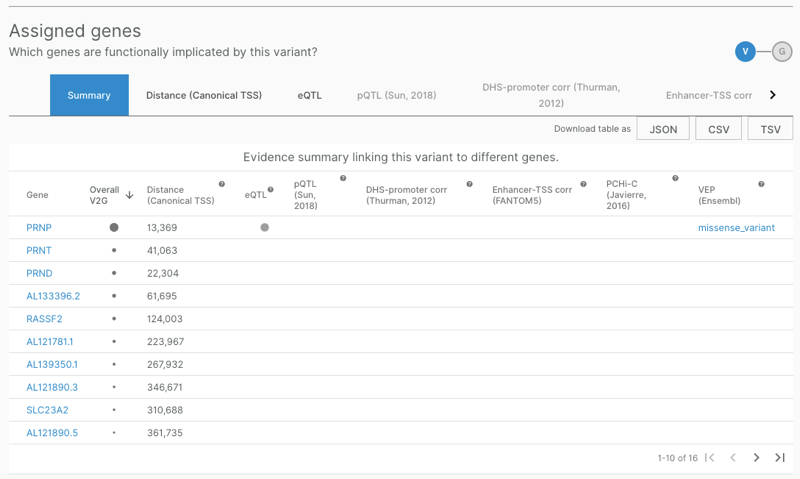
After browsing these data for several of the PRNP GWAS hits in OTG, I did not identify any phenotypes besides prion disease where PRNP was among the top three implicated genes. In contrast, for the prion disease-associated lead SNP from [Mead 2009], 20_4699605_A_G, PRNP was the clear top-ranked gene based in its VEP annotation and its eQTL status:
To check in a systematic way the intuition that the above examples gave me, I wanted to look at the evidence linking all 59 GWAS hits to PRNP. I set out to evaluate the level of v2g evidence for the 32 unique SNPs that accounted for the 59 GWAS hits listed in OTG. I manually downloaded all 32 v2g files in order to summarize them and mash them up with the Associated Studies table.
A summary of these data (Table 1) reveals that only the two SNPs located physically in PRNP and associated to prion disease (bold rows) show strong evidence for assignment to PRNP, with overall_v2g scores that comprise 28.7% and 40.0% of the total for all genes. For all other reported associations, PRNP’s share of overall_v2g score is <10% of the total. Indeed, for the SNP (20_4176425_C_G) that has both the largest number of associations (N=16) and the highest-listed (#1: immature reticulocyte fraction in UK BioBank) PRNP’s share of overall v2g is 0.0%.
| variant | bp to PRNP TSS | N hits | GWAS hit phenotypes | PRNP v2g share |
|---|---|---|---|---|
| 20_3355571_C_CA | -1,330,779 | 1 | High light scatter reticulocyte count | 0.0% |
| 20_3617227_T_C | -1,069,123 | 1 | Breast cancer | 0.0% |
| 20_3632959_G_A | -1,053,391 | 1 | High light scatter reticulocyte count | 0.0% |
| 20_4122307_G_A | -564,043 | 2 | Trunk predicted mass, Trunk fat-free mass | 0.0% |
| 20_4139131_C_T | -547,219 | 4 | Reticulocyte count, Reticulocyte fraction of red cells, High… | 2.5% |
| 20_4139576_G_A | -546,774 | 3 | Reticulocyte count, Reticulocyte percentage, Immature reticu… | 2.4% |
| 20_4145210_C_T | -541,140 | 1 | High light scatter reticulocyte count | 0.0% |
| 20_4148407_G_T | -537,943 | 1 | Reticulocyte count | 3.9% |
| 20_4162192_G_A | -524,158 | 1 | Haemoglobin concentration | 0.0% |
| 20_4176425_C_G | -509,925 | 16 | Reticulocyte count, Immature reticulocyte fraction, Mean cor… | 0.0% |
| 20_4180853_C_T | -505,497 | 1 | White blood cell count | 0.0% |
| 20_4182655_A_G | -503,695 | 1 | High light scatter reticulocyte count | 0.0% |
| 20_4188432_G_A | -497,918 | 1 | Immature fraction of reticulocytes | 0.9% |
| 20_4189582_G_C | -496,768 | 1 | Mean reticulocyte volume | 0.9% |
| 20_4222579_C_T | -463,771 | 2 | Heel bone mineral density, Heel bone mineral density | 2.5% |
| 20_4262797_G_C | -423,553 | 1 | polycystic kidney | Non-cancer illness code, self-reported | 2.4% |
| 20_4293964_T_G | -392,386 | 1 | Lymphocyte count | 4.3% |
| 20_4445885_G_A | -240,465 | 3 | hypertension | Non-cancer illness code, self-reported, High … | 9.4% |
| 20_4599109_G_A | -87,241 | 1 | High light scatter reticulocyte percentage | 9.4% |
| 20_4696446_G_A | 10,096* | 2 | Creutzfeldt-Jakob disease (variant), Creutzfeldt-Jakob disea… | 28.7% |
| 20_4699605_A_G | 13,255** | 1 | Creutzfeldt-Jakob disease | 40.0% |
| 20_4721739_G_A | 35,389 | 1 | Dark brown | Hair colour (natural, before greying) | 8.9% |
| 20_4795437_C_T | 109,087 | 1 | DNA methylation variation (age effect) | 7.5% |
| 20_4805434_T_C | 119,084 | 1 | Breast cancer | 6.2% |
| 20_4999759_C_T | 313,409 | 1 | Red cell distribution width | 3.1% |
| 20_5002446_G_A | 316,096 | 3 | Mean corpuscular haemoglobin, Mean corpuscular volume, Mean … | 3.2% |
| 20_5004696_A_T | 318,346 | 1 | Mean corpuscular volume | 3.3% |
| 20_5073907_C_G | 387,557 | 1 | Mean platelet (thrombocyte) volume | 1.7% |
| 20_5128756_T_C | 442,406 | 1 | Comparative height size at age 10 | 1.2% |
| 20_5132068_T_A | 445,718 | 1 | Standing height | 1.2% |
| 20_5493266_C_A | 806,916 | 1 | Breast cancer | 0.0% |
| 20_6858847_A_G | 2,172,497 | 1 | Breast cancer | 0.0% |
Table 1. Summary of variant-to-gene (v2g) evidence for PRNP GWAS hits in Open Targets Genetics.Variants are in GRCh38 coordinates. TSS = transcription start site. “v2g share” indicates the overall_v2g score for PRNP divided by the sum of that for all genes. *PRNP intron variant. **PRNP missense variant.
Visualizing the same results from Table 1, with genome context included to show other genes, makes the differences even more obvious (Figure 1).
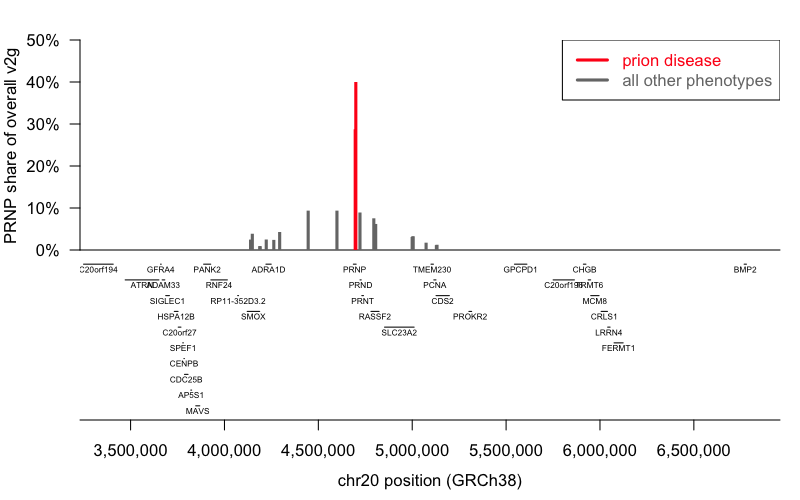
Figure 1. PRNP share of overall v2g score by genomic position. Data from Table 1 as a barplot (top) colored by phenotype, overlaid with genomic context (bottom).
Remember that all of the above analyses were based on variant-to-gene (v2g) data, meaning, functional evidence for the variant, independent of the context of its LD peak in the original GWAS. What about full colocalization analysis? Back on the PRNP gene page, below the Associated Studies table appears the Colocalising Studies table, listing SNPs for which full GWAS summary statistics have been examined and found to colocalize with GTEx eQTLs:
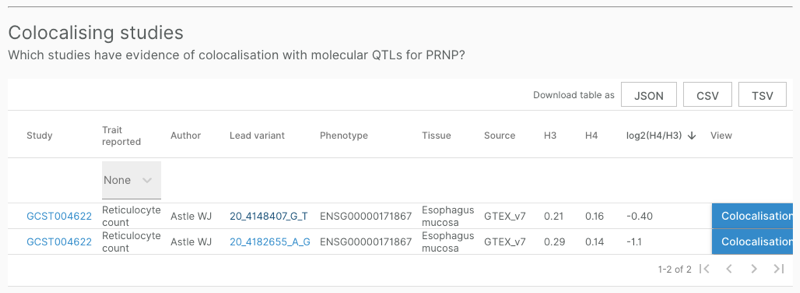
This table contained only two variants, so it could not explain all of the GWAS hit assignments that appeared above in the Associated Studies table. Still, I wanted to take a closer look. Clicking on the “Colocalisation” button leads to a different page, specific to the combination of SNP and study, such as that for 20_4148407_G_T in [Astle 2016]. This page features a table, QTL Colocalisation, that is conceptually similar to the v2g table above:
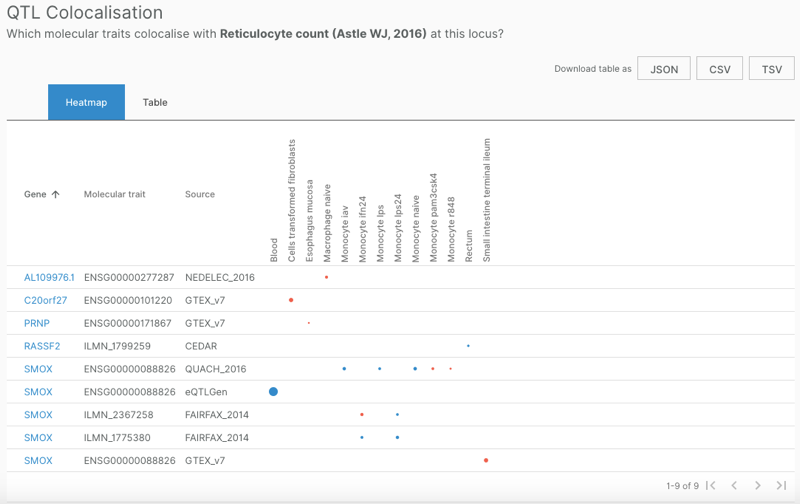
There is one row per combination of gene, molecular trait, and source of evidence, and one column per tissue. Here, the dots are not only sized, but also color-coded: those where the gene is more likely than not to be causal (based on posterior probabilities) are blue, while those more likely to be non-causal than causal are red. Here, again, SMOX is the overwhelmingly implicated gene. PRNP has only a small red dot in one tissue, indicating that while there’s not no evidence that PRNP could be functionally related to this SNP, it is more likely unrelated than related. The one other GWAS hit with colocalization data, 20_4182655_A_G, likewise showed PRNP as a red dot, indicating it is probably not the causal gene.
In both of those cases, the limited evidence in favor of PRNP was reported to come from GTEx expression data in one tissue, esophageal mucosa. These two “colocalized” SNPs were both located some 500+ kb upstream of PRNP, whereas I hadn’t been aware that GTEx had identified any eQTLs for PRNP that far upstream. This made me curious to see what the actual colocalization of the LD peaks looked like. OTG does not display the overlapping LD peaks, so I went back to the raw data, downloading the GWAS summary statistics for reticulocyte count from [Astle 2016] (specifically ret_N170641_narrow_form.tsv.gz linked from here) and the GTEx v8 eQTL data for all genes in esophageal mucosa (Esophagus_Mucosa.v8.signif_variant_gene_pairs.txt.gz from this file from here). Mashing up these two datasets revealed no visually obvious evidence for colocalization, nor even evidence of any nominal overlap among associated SNPs (Figure 2).
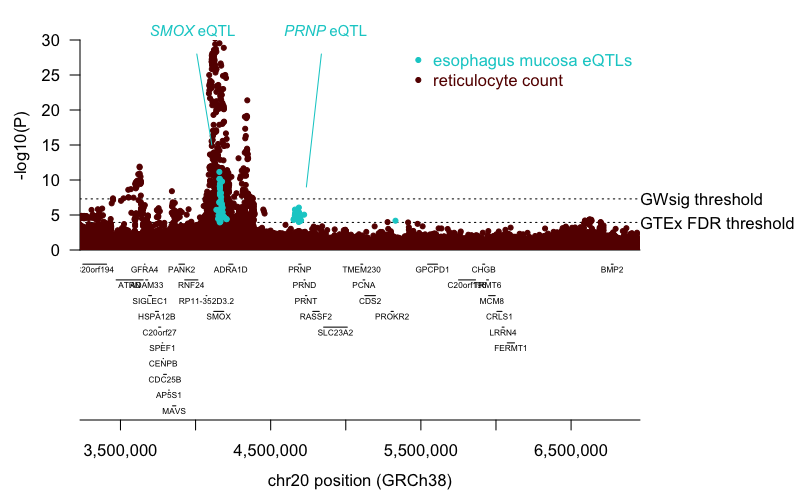
Figure 2. Association peaks for esophageal mucosa gene expression and reticulocyte count.Variants with qval < 0.05 from GTEx v8 in esophageal mucosa, for any gene (cyan), versus variants tested for association with reticulocyte count in [Astle 2016] (maroon).
Indeed, message that strikes me visually from this plot is the near-perfect colocalization between the reticulocyte count LD peak and the peak for the SMOX eQTL. The PRNP eQTL is clearly distinct and does not overlap at all with the genome-wide significant peak for reticulocyte count. How, then, was this considered a colocalization hit for PRNP? I don’t have a full explanation, but from my limited understanding, colocalization analysis uses all SNPs and not only genome-wide significant SNPs, so it could be that some subtle whiff of signal underneath the genome-wide threshold, perhaps a similarity in the shape of the curves, near PRNP, led to a false positive here.
Conclusions about PRNP
From all of the above analyses, I concluded that there is no compelling evidence that PRNP is associated in GWAS to any human phenotypes other than prion disease at the present moment.
Evidence for involvement of PRNP in hematologic phenotypes from animal models is similarly scant, and results can likewise be influenced by linkage. A macrophage phenotype was once reported in PrP knockout mice [de Almeida 2005], but was later demonstrated to be a linkage artifact [Nuvolone 2013, Nuvolone & Hermann 2016, Nuvolone 2017] due to polymorphisms in Sirpa, 2.4 Mb upstream on mouse chromosome 2, that segregated with the Prnp knockout allele even after 12 generations of backcrossing. One study has reported a red cell count phenotype in PrP knockout goats [Reiten 2015], but even in nominal terms, without applying multiple testing correction for all the phenotypes measured in that paper, the association was only suggestive (P = 0.08). Even if that trend is real, it could be due to linkage: the goat ortholog of SMOX is 8 Mb downstream of PRNP on Capra aegagrus hircus chromosome 13, plenty close enough for strong linkage within the single goat herd where the naturally occurring PrP knockout allele was first reported just seven years ago [Benestad 2012].
Conclusions about gene assignment in GWAS
What all of this drove home for me is that assigning causal genes to GWAS hits is really hard! A number of my colleagues at the Broad Institute do research in this area, and now I have a better understanding of what they are up against. On one hand, it’s clear that we cannot afford to just assume that the gene that contains or is nearest to the most significant SNP is the causal gene — this will lead to a tremendous rate of both false positives and false negatives. At the same time, the best methods available for assigning causal genes based on functional genomics datasets can ultimately only give probabilistic answers, which leads to a need to set some minimum threshold of evidence to call a gene as causal or potentially causal, and so there is a sensitivity/specificity tradeoff there too. In the Open Targets Genetics example above, the aggregation and visualization of v2g data is fantastic, but the threshold of evidence required for including a GWAS hit on a gene’s page appears to be fairly permissive, meaning the user may want to inspect the hits carefully to decide if they are really associated to the gene of interest. Having on hand some of the context — how many other genes could this SNP be associated to? what fraction of the evidence supports my gene as opposed to others? — can be very helpful. Ultimately, deep visual inspection of the raw data supporting an association, as in the colocalization example above, may convince you that a gene assignment is a clear false positive, even though the algorithm that generated that assignment has been deeply vetted and QC’ed in the aggregate.
Why is all this so hard? In some sense, it is because we are up against the fact that the genome is large. Mark Daly once pointed out that the advent of GWAS meant the advent of QC:
It soon became clear that GWAS were different from anything that had come before them in molecular biology. In all of molecular biology up to that point, you got a gel or some sort of readout that you could look at with the naked eye and decide whether you believed the data. With GWAS you could now get a volume of data that no one human would ever look at with the naked eye… Thus, we had to develop technical QC… genetic QC… analytical QC… Those QC efforts were 95% of the labor.
Over time, there’s no doubt that the underlying datasets, the algorithms, and the way the data are filtered and presented, will all continue to improve, and I imagine that artifacts like the PRNP colocalization hit shown above will gradually be weeded out. But weeding them out on a genome-wide scale is a really hard task, and until then, there’s no substitute for a deep dive on the gene you care most about.
Data and R source code to reproduce the analyses in this post are available on GitHub.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.