The anticipation parallelogram
Last month, I was excited to see a study [Cohen 2019] that agrees with something I concluded several years ago [Minikel 2014], while providing interesting additional lines of evidence.
After I began re-training as a scientist eight years ago, the first research study I led looked at a phenomenon called anticipation [Minikel 2014]. In genetics, anticipation is when the age of onset of a particular genetic disease gets younger and younger in each new generation of a family. Anticipation was recognized in myotonic dystrophy, among other diseases, a century ago [Adie 1923]. The concept fell out of favor after it was shown that it could result simply from biased case ascertainment [Penrose 1948], but then re-emerged in the 1980s and 1990s when several genetic diseases, including myotonic dystrophy and Huntington’s disease, were shown to be caused by unstable expansions of trinucleotide repeat DNA. The pathogenic repeating segment of DNA could get longer in each generation, accounting for the earlier onset of disease [Harper 1992].
Today, there’s no doubt that anticipation is unfortunately a real phenomenon in some diseases. But a series of studies proposing anticipation in genetic prion disease [Rosenmann 1999, Mitrova 2012, Pocchiari 2013] seemed hard to believe. Unlike myotonic dystrophy or Huntington’s disease, prion disease is not caused by a trinucleotide repeat expansion. (About 10% of genetic prion disease cases are caused by extra octapeptide repeats [Minikel 2016], which must have arisen from an unstable DNA repeat expansion at some time in history, but expansion of the repeat from generation to generation within a family has not yet been documented, and in any case, none of the anticipation studies focused on octapeptide repeat mutations — all of them were focused on the most common genetic mutation, E200K.) How could this point mutation result in anticipation?
My 2013 self hypothesized that the supposed anticipation might just be an artifact. Age of onset in genetic prion disease is highly variable, so by chance you’ll have some parent-child pairs where the parent has a relatively older age of onset and the child has a relatiely younger age of onset, and some pairs where the opposite is true. But one of those types of pairs is much more likely to appear in a researcher’s dataset than the other:
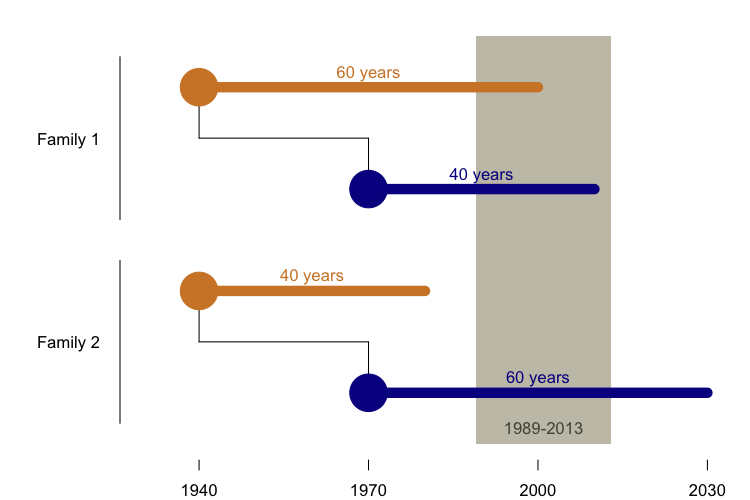
The reason is that before 1989, we didn’t have the ability to sequence the PRNP gene and barely anyone was studying genetic prion disease, so we have very little ability to identify cases before then. And because many people don’t get predictive genetic testing, we have relatively little insight into the potential cases whose onset still lies in the future. That leaves us just a 30-year historical window in which to observe disease onsets. Many children were born 30 years later than their parents, so in order to be identified and included in a dataset of parent-child pairs who both died of genetic prion disease, it is virtually a pre-requisite that the child have a younger onset than the parent. So in the above diagram, Family 1 is likely to appear in our dataset, and Family 2 is not. We showed through simulations that this phenomenon was sufficient to explain the reported “anticipation” in genetic prion disease. Then, using data from four clinical centers that had studied families with the E200K mutation, we showed that the datasets bore all the signatures of ascertainment bias that were predicted by our model, that this was sufficient to explain the previously reported results, that the problem could be partly mitigated by performing a survival analysis including asymptomatic mutation carriers, and that even non-prion disease deaths in the same families showed the same pattern of earlier deaths in subsequent generations [Minikel 2014].
The new study [Cohen 2019] terms the ascertainment bias problem a “rhomboid shaped artifact”, referring to what happens when you plot year of birth versus age of onset. What does that mean, exactly? Here’s what the geometry looks like using our dataset from a few years ago [Minikel 2014]:
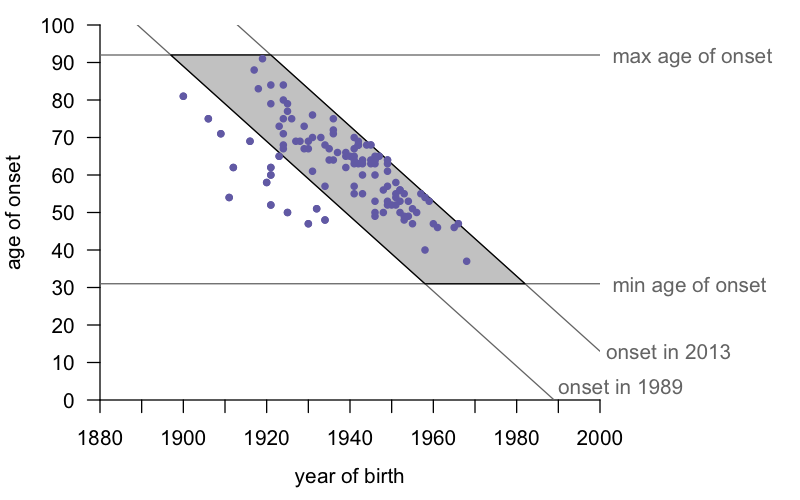
For simplicity, I’d just call this a parallelogram. (A rhomboid, the term the authors used, is a parallelogram of unequal sides, not to be confused with a rhombus, which is a parallelogram with all four sides being of equal length.) In any case, the insight is this: if one draws lines corresponding to the minimum amd maximum ages of onset (for the E200K mutation, these are 31 and 92 according to our most recent study [Minikel 2019]) and the minimum and maximum years of ascertainment (1989 and 2013 for our 2014 study), one finds that virtually all the data points fall inside the resulting parallelogram. The handful of cases to the left are people who died before the E200K mutation was known, yet whose disease we feel we can confidently identify as genetic prion disease based on family history. There are a handful of such people, but they aren’t enough to overcome the bias in the rest of the data. We basically face the following constraint: a person born in 1960 needs to die pretty young in order to get counted, and their parent born in 1930 needs to die pretty old in order to get counted. It’s not that there aren’t people born in 1930 with the mutation who had an unlucky early onset, they just probably never got diagnosed or studied. And it’s not that there aren’t people born in 1960 with the mutation who won’t live to a good old age, they’re just still alive today, so we don’t yet know their age of onset.
The new study [Cohen 2019] shows a similar diagram and makes a similar point, using a completely separate dataset of 266 people in 73 families in Israel. But they also make two interesting arguments that we didn’t make in 2014, that bolster the case even further.
First, whereas the individuals in our 2014 dataset were from dozens of unrelated families across the U.S., Europe, and Australia, it is believed that all or nearly all E200K individuals in Israel are descdended from a single founder. The authors therefore argue that if anticipation were real, then age of onset should be dropping over time, so that cases diagnosed in 2015 should be younger than those in 1995, because people are on average one more generation removed from that original founder. Instead, when they plot age of onset versus year of onset, they find that age of onset is pretty steady over time. If anything, age of onset has gone up just slightly, probably because of better diagnosis of the disease in older individuals.
Second, they use sporadic prion disease patients as a sort of negative control — since these individuals have no mutation, there should be no opportunity for anticipation. And yet, in sporadic prion disease patients in Israel, they find the same trends — the years of birth and ages of onset are inscribed within the same parallelogram, and the flat or just slightly increasing relationship between age of onset and year of onset is similar.
What does all this mean for patients? Age of onset is always a hard topic — the truth is that age of onset in genetic prion disease is highly variable and as far as we can tell, none of the usual factors seem to predict it, including family history [Minikel 2019]. There might be a large element of true randomness, which is hard to accept, because after testing positive for a mutation, no one feels lucky. But the good news is, at least age of onset is not getting younger over time or over generations, and there is no reason to expect that your onset will necessarily be younger than your mother’s or father’s was. But if that alone is not enough cause for optimism for you, don’t worry, it’s not enough for us either. Our quest for a cure races on.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.