Details emerge on tominersen trial halt
This is an update for the prion disease community on tominersen, an ASO drug for Huntington’s disease that was in a Phase III trial called GENERATION HD1, for which dosing was stopped last month following an independent data monitoring committee (iDMC) recommendation. Tominersen was invented by Ionis Pharmceuticals and clinical development was being led by their partner Roche. Because Ionis Pharmaceuticals is developing an ASO drug for prion disease, many in our community have been watching the tominersen results with great interest, and last month’s press release did not give any details on what happened in the trial. At the CHDI Annual HD Therapeutics Conference yesterday, Roche’s Dr. Scott Schobel gave a detailed presentation on everything they know so far about the trial’s preliminary results. The host, CHDI’s Chief Scientific Officer Dr. Robert Pacifici, stated that recordings of all the talks will be posted publicly, and no ban was imposed on social media sharing of presentations — so I’m blogging the talk here to share and discuss with our community.
rationale for dosing regimen
Dr. Schobel opened by reviewing the rationale for the exact dosing regimens chosen for study in Phase III. This included some details worthy of recounting here, because while they have been previously discosed publicly, they were not entirely obvious to me from reading the published work on tominersen.
In the Phase I trial [Tabrizi 2019], participants received 4 doses spaced 4 weeks apart. 4 weeks after the final dose (t=20 weeks), mutant huntingtin (mHTT) in CSF had been lowered ~40% in the highest two dose groups, 90 mg and 120 mg. Participants in that trial went on to an open-label extension trial (dubbed OLE-CS2). Data from OLE-CS2 have not been published but the results turned out to critically inform design of the Phase III trial, on two dimensions: dose level, and dosing frequency.
The Phase I trial observed no dose-limiting toxicity. There was only one serious adverse event and it was in the placebo group; less serious adverse events were distributed evenly between placebo and active drug groups and generally did not appear drug-related. The one potential concern was that, after the final dose, the 90 and 120 mg groups had some rise in the neuronal damage marker CSF neurofilament light (NfL), and in brain ventricular volume:
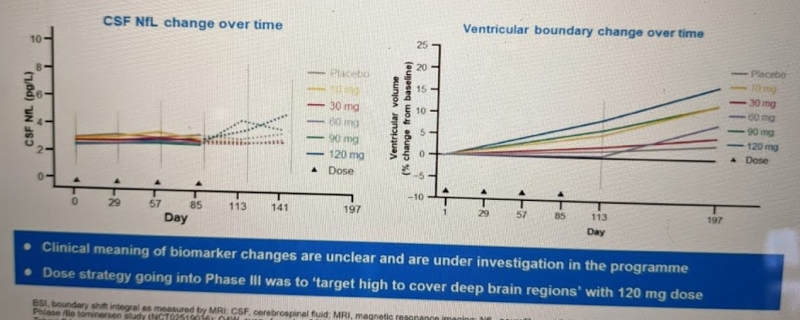
However, they ended up deciding these changes were not a big enough concern to limit dose level, for three reasons. First, these are just fluid/imaging biomarkers, with no clear clinical meaning and no observed impact on patient phenotypes. Second, after the Phase I ended, they found that these changes reversed upon cessation of dosing [Tabrizi 2019]:
By the start of the extension study (7 to 27 months after the final doses were administered in this trial), the concentrations of neurofilament light protein in the CSF had generally returned to pretrial concentrations.
Third, in OLE-CS2, they found that the NfL increase replicated when dosing began, but then spontaneously reversed itself despite continued dosing:
During the extension study, the concentrations rose with a time course and magnitude that were similar to those observed in this trial and then decreased at later time points despite continued treatment (unpublished data).
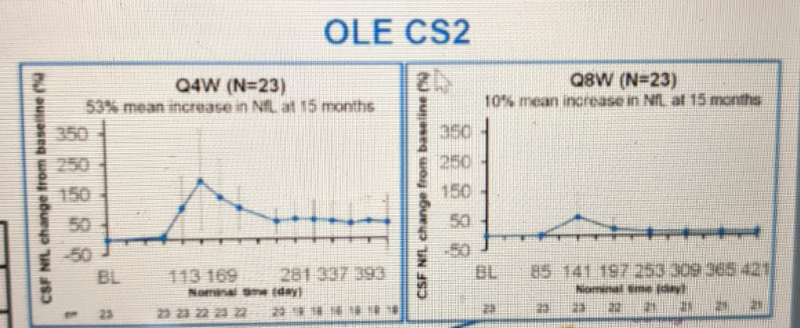
Based on all the above considerations, and a desire to make sure they achieved drug distribution to the deep brain regions (especially striatum) that are most impacted in HD, Roche decided to proceed with the 120 mg dose in Phase III.
Then there was the question of dosing frequency. 4 doses at 4-week intervals had lowered mHTT 40% in the Phase I, but it turns out OLE-CS2 revealed this was not at all the limit of potency that the drug could achieve. Patients who stayed on 120 mg every 4 weeks (Q4W) dosing eventually got to a 70% reduction in mHTT by t=15 months. However, this frequency of dosing was not as well tolerated over this longer time period: there were more adverse events — and so a significant fraction of patients missed some doses. OLE-CS2 also had patients on an every 8 week (Q8W) dosing regimen and this led to 44% reduction in mHTT by t=15 months:
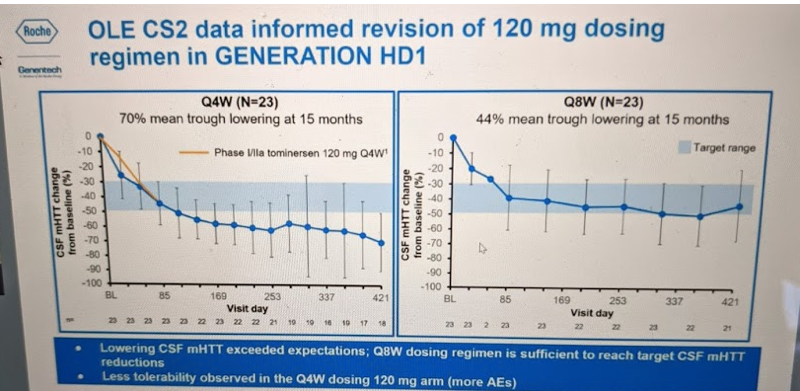
They therefore amended the Phase III trial protocol to examine two dosing frequencies: Q8W and Q16W. The Q16W regimen had not yet been tested in any previous trial, but the preclinical animal data had suggested that the drug would largely wash out by 16 weeks post-dose. If so, then the Q16W regimen might test the hypothesis that periodic, rather than sustained, huntingtin lowering could be beneficial — the so-called “huntingtin holiday” hypothesis.
interim efficacy evaluation
Dr. Schobel began discussion of the GENERATION HD1 results by emphasizing how preliminary they are. First, the data freeze that the iDMC examined in order to make the decision to stop the trial was from Feb 5, 2021, but the iDMC needed time to compile and analyze the data and make a recommendation, so the final decision to stop dosing in the trial did not occur until Mar 22, 2021. As a result, the data freeze on which this presentation is based includes data on ~60% of participants through the t=17 month mark, but another ~10% of participants reached the 17 month mark before dosing stopped. Therefore, by the next time you see a presentation or publication on these data, the numbers will certainly have shifted. Second, the data available right now are limited to scales and scores measuring patients’ functional/motor/cognitive change, as well as brain imaging data. There are not yet any data from fluid biomarkers such as mHTT or NfL, or drug concentrations in CSF.

With those caveats, Dr. Schobel presented data from the primary and secondary endpoints that are available so far. This trial (NCT03761849) had two registered primary endpoints: the Composite Unified Huntington’s Disease Rating Scale (cUHDRS), and total functional capacity (TFC) score, as well as a host of secondary endpoints. He showed the two primary endpoints first. Here is what cUHDRS looked like:
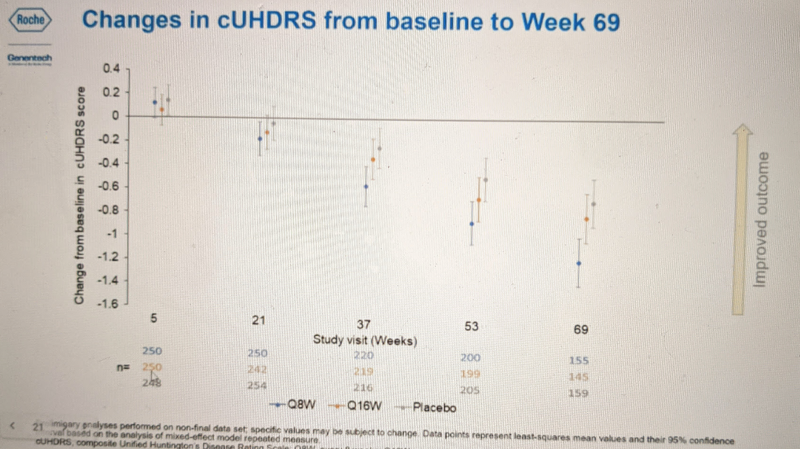
Blue is the more frequent dosing (Q8W) group, orange is the less frequent dosing group (Q16W), and gray is placebo. The bars represent 95% confidence intervals. These are patients with manifest Huntington’s disease, and the natural history of the disease absent an effective therapeutic is for cUHDRS to decline over time, meaning patients get worse. And indeed, all groups including the placebo group got worse over time. By 69 weeks, the Q8W group was significantly worse than placebo (non-overlapping confidence intervals). None of the other timepoints appear to reach statistical significance, but if there exists any trend here, it is for cUHDRS scores to decline slowest in the placebo group, slightly faster for Q16W, and faster still for Q8W.
For TFC, none of the group-wise differences were significant — all confidence intervals overlap — but the trend appears broadly similar:
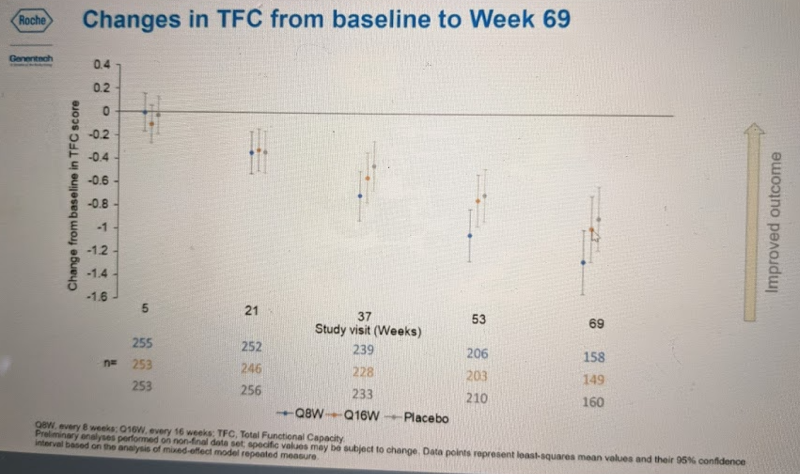
Dr. Schobel then showed similar plots from the subset of secondary endpoints for which data are available: total motor score (TMS), symbol digit modalities test (SDMT), Stroop word reading (SWR), clinical global impression (CGI-S), and others which I missed while furiously taking notes. For most of these endpoints, no differences between placebo and drug-treated groups were significant, but wherever there existed a trend, it was the same as above — generally, Q8W seemed to do at least a bit worse than placebo, and Q16W was either somewhere in between or was spot-on with placebo.
At the end of this section of the talk, Dr. Schobel summarized the findings as follows: there was “no evidence for patient benefit,” and the Q8W regimen had an “unfavorable efficacy signal vs. placebo” while Q16W was “overlapping with placebo”.

Next he transitioned to the safety evaluation.
interim safety evaluation
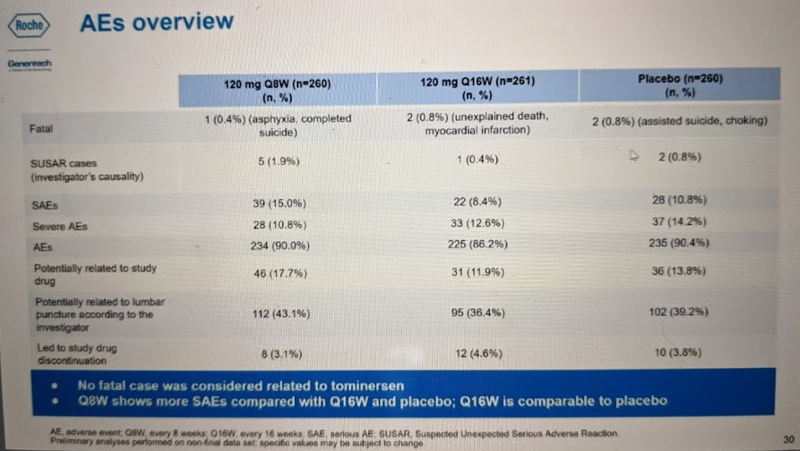
The safety data from GENERATION HD1 so far can mostly be summed up in one table, below. There was no difference in fatalities or trial discontinuation between the drug and placebo groups, and no difference in adverse events overall. There were no differences in suicidal ideation. The two categories with slightly unfavorable signals were SUSAR (“Suspected Unexpected Serious Adverse Reaction”, i.e. serious adverse events deemed by the study investigator to be drug-related), and SAE (“serious adverse event”). SUSARs affected 1.9% of participants in the Q8W group and 0.4-0.8% in the other groups. SAEs affected 15% of Q8W participants and 8.4-10.8% in the other groups.
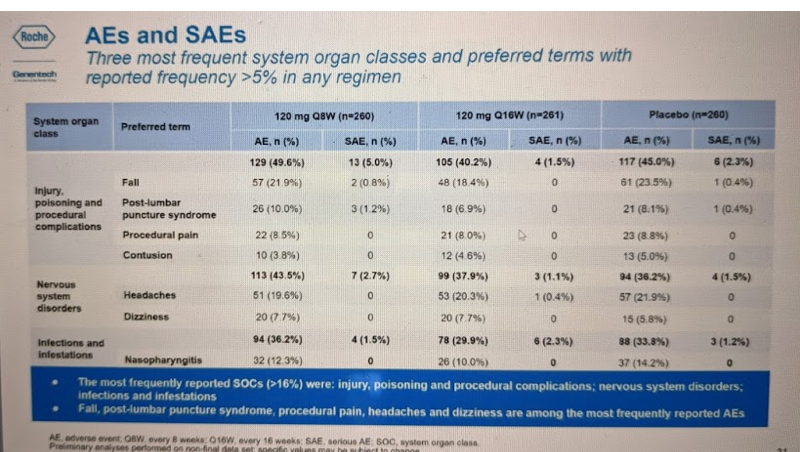
Most of the AEs and SAEs fell into the “other” categories (preferred term = blank):
The other clearly significant difference between groups in this trial was in the ventricular volume imaging outcome, see the right panel of this slide:
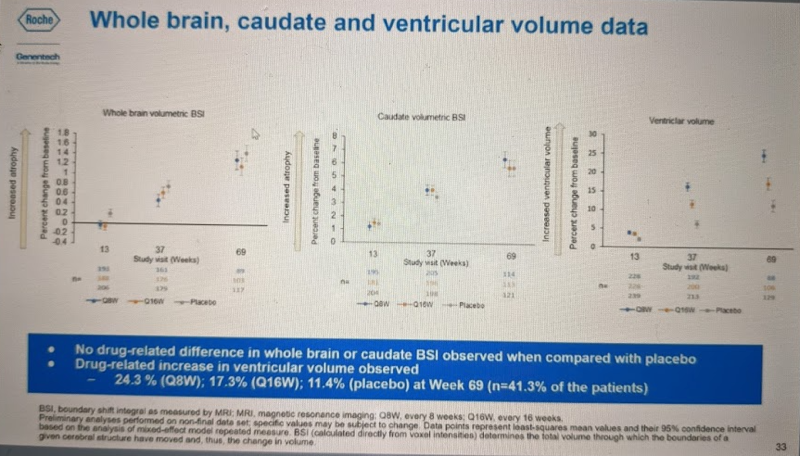
Again, these are manifest HD patients, so the natural history is for ventricular volume to expand over time as the brain parenchyma atrophies, and we see this in the placebo (gray) group. The Q16W (orange) group had significantly worse increase in ventricular volume than placebo, and Q8W (blue) group was worse still. It is not clear, however, what explains this change. If you look at the other two panels, you can see that the volume of whole brain parenchyma (far left) and caudate in particular (middle) atrophied in all groups as expected for HD progression, but was no different in drug vs. placebo groups. Therefore, these data do not suggest that the exacerbated increase in ventricular volume on drug is due to exacerbated brain atrophy.
Instead, one possibility is that some percentage of the time, tominersen may cause some degree of hydrocephalus, a treatable condition of having too much CSF. Across all the tominersen trials with data available so far there have been 5 symptomatic cases of hydrocephalus, tending to occur in higher dose groups. Hydrocephalus occurred in 4/636 people who have ever been on the Q8W regimen. Another 16 asymptomatic cases have been detected. Overall, then, there may be a spectrum of CSF volume increase in people on frequent dosing of tominersen, with a handful of cases rising to the level of symptomatic:

In the Q&A, host Dr. Robert Pacifici noted that a handful of cases of hydrocephalus have also been reported for the approved ASO drug nusinersen, for spinal muscular atrophy. This was in post-marketing reports to regulators, not in controlled trials, so we don’t know for sure that it’s drug related. The FDA adverse event reporting system (FAERS) is not set up to easily make cumulative queries of these kind of reports, but I quickly downloaded and checked just the most recent quarter (2020Q4) for which data are available, and there were no cases of hydrocephalus on nusinersen reported in that quarter, despite thousands of patients on drug. So this event seems to be quite rare. Nonetheless, FDA did add a warning about this potential side effect to nusinersen’s label in 2018:

Dr. Schobel also briefly touched on CSF total protein and white blood cell count, the only two CSF lab tests for which results are available — again, mHTT and NfL measurements are still pending. For both of these metrics, elevation out of the reference range was a bit more common in the Q8W group:
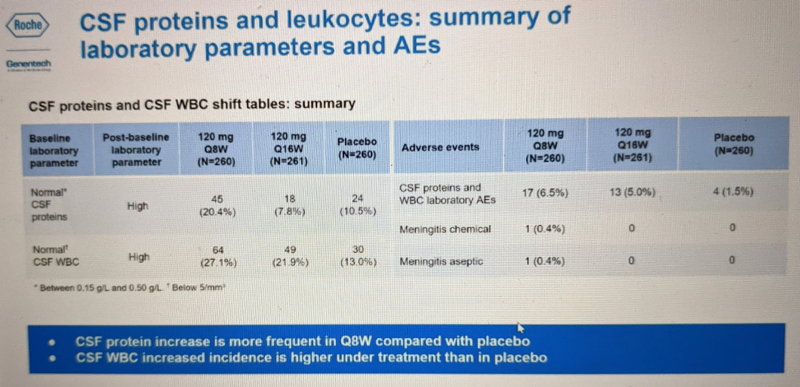
These metrics do not have any obvious clinical implication and were not discussed further. They also looked at so-called “class risks” such as liver and kidney function and clotting. No concerns were identified in any of these areas:

These are presumably being called “class risks” because these are the kind of things where there exists a potential for a “class effect”, where a class of drugs — here, ASOs — could share a common toxicological liability. Many drugs are metabolized through kidney and liver and could exhibit toxicity there. And a few ASOs are associated with increased risk of thromocytopenia (low platelet count leading to a potential lack of blood clotting) — for example, the ASO volanesorsen, for hypertriglyceridemia, is approved in the E.U. but not the U.S. in part because FDA was concerned about thrombocytopenia. However, Ionis’s own analysis of its clinical trials database suggested this is not a generalizable side effect across all ASOs — only 3/16 ASOs that have been dosed into humans had a >10% incidence of >30% decrease in platelet counts [Crooke 2017].
Overall, the Q8W dosing regimen had a higher rate of adverse events and was less well tolerated than the Q16W and placebo:

next steps
Importantly, Dr. Schobel emphasized that while dosing is stopped in GENERATION HD1, the trial will go on, meaning that participants will be asked to continue to make study visits and be evaluated for safety signals, outcome measures, imaging, CSF biomarkers, and so on. This is absolutely critical, because it will allow the evaluation of longer-term outcomes, and in particular, will make it possible to determine whether any changes that occurred on study drug are reversible. For example: does ventricular volume go back to being indistinguishable from placebo after drug administration is stopped?

Dr. Schobel also emphasized again that these results are preliminary on several dimensions, and that having the full dataset will be critical to understanding what exactly happened in this trial. This part of the talk was very compelling: he described several hypotheses that can be explored using the data that will eventually be available from this trial. For example, does disease stage affect outcomes? He admitted that our ability to interrogate this is limited by the fact that premanifest subjects, in whom some hypothesize the greatest benefit of huntingtin lowering might be achieved, were not included in the trial. Nonetheless, about half the participants were in the early stages of HD symptoms (phase I) while about half were a bit more advanced, in stages II-III. Post-hoc subgroup analyses may be able to at least give a suggestive answer to the question of whether the unfavorable changes seen overall were driven more by people with more advanced or less advanced disease. Also, while people were only on two different dosing regimens, about 35% of people missed at least one dose and 10% missed 2+ doses (mostly due to COVID-19), plus, even among those getting the same dose, the response in terms of mHTT lowering may not be identical. Thus, in time it will possible to ask whether total drug exposure or target engagement (mHTT lowering) are correlated with clinical outcomes. Later, in the Q&A, he also noted that of the individuals who passed away during the trial, one individual who was on drug agreed to donate their brain, so there will eventually be autopsy data available. This will make it possible to quantify drug concentration in brain parenchyma and ask questions like whether atrophy was observed in the regions with highest drug concentration.

He emphasized that while the trial was not a success, there is a lot to be learned here and that Roche plans to share these learnings with the community:
interpretation
In my previous post on this topic, I speculated on several possibilities of what happened in this trial. While the data presented yesterday are far from final or definitive, we can now begin to refine the list of possibilities just a little bit.
When the trial was stopped, one possibility that stood out in our minds was “futility,” meaning that perhaps dosing was stopped simply because there was no hope of the drug candidate proving effective. We can now rule that out: while not all differences were significant, the overall picture appears to be that the Q8W dosing regimen was actually worse than placebo. This is still not quite the worst case scenario: in my previous post I mentioned the example of the experimental drug revusiran, where significantly more patients died on drug than on placebo. At least, tominersen doesn’t seem to have caused excess deaths. Still, it’s a very bad trial outcome. That in turn focuses the list of possible explanations a bit. If the trial had simply been futile, one possible reason would be if the drug did not achieve adequate distribution in the brain. To be sure, we still don’t have any brain distribution data, and we certainly can’t prove that the drug did reach active concentrations across the brain. But, the problem in this trial was clearly more than just a lack of drug distribution, and indeed, if anything, the adverse outcome associated with frequent dosing is at least suggestive that the drug did reach the brain in significant concentrations.
The fact that cUHDRS got worse on the Q8W regimen does not necessarily mean that tominersen made HD worse. It’s important to remember that the trial was designed around seeing an improvement in HD, so all the endpoints are HD-related. If someone had bothered to assess, say, a scale developed for Alzheimer’s or Parkinson’s such as MMSE or Hoehn & Yahr, it’s possible those scales would have also showed people on drug worsening, simply because there is nonzero overlap in the symptoms that occurred in the drug group, and the symptoms of AD or PD, but of course that wouldn’t mean the drug “gave people AD” or “gave people PD”. In other words, basically any neurological effect of the drug is going to show up in those HD scales such as cUHDRS, even if the effect has nothing to do with HD, simply because those scales are what were measured in the trial.
Still, people on drug did worse, overall, than people on placebo. That leaves us with the key question of whether the unfavorable effect was an on-target or off-target outcome. In other words, was it result of huntingtin lowering, or some other effect of the drug?
I’m an outsider to the HD field, and not nearly as well-read on it as I am on prion disease. But from what I see, huntingtin lowering is a therapeutic hypothesis with sound scientific foundations and fairly broad acceptance in the HD scientific community†. Nonetheless, lowering the protein that causes the disease is not quite as straightforward a proposition in HD as it is in prion disease. Huntingtin knockout is embryonic lethal in mice [Duyao 1995], and hypomorphic mutations in huntingtin appear to cause a severe recessive disease in humans [Rodan 2016]. So there is definitely such a thing as too little huntingtin. If (and it’s a big if) the problem in this trial was on-target, then the fact that the Q16W regimen was no better than placebo (and on some metrics slightly worse) may suggest that there is either a rather narrow therapeutic window, or no therapeutic window at all, for non-allele-specific huntingtin lowering. If so, allele-specific lowering could be the only route forward. There are drug candidates in the pipeline for that: as noted in my last post, Wave recently dropped two allele-specific drug candidates that failed in Phase I for lack of potency, but has a third candidate advancing towards trials, and Ionis has developed allele specific ASOs against HTT as well [Southwell 2014, Southwell 2018]. For what it’s worth, both Roche and Ionis have said they are not done with huntingtin lowering, and Dr. Schobel reiterated his belief in the therapeutic hypothesis, as did other investigators who spoke at HDSA’s webinar on tominersen last month. Dr. Pacifici noted that while a positive result in this trial would have proven the therapeutic hypothesis, a negative result does not refute it.
†I’ve occasionally heard the suggestion that HD could arise from a dominant negative gain-of-function that abrogates the activity of the wild-type allele, in other words, that the disease actually arises from a loss of HTT function. But this view does not seem to be widespread, and anyway, heterozygous knockout of HTT does not appear to cause severe or highly penetrant disease in humans or mice, so even if that were true, specific silencing of mutant huntingtin would remain a viable strategy.
The other possibility is that the effect was off-target. If so (and again, that’s a big if), I see two key questions emerging. First, is this a property of this one ASO, or a class effect that could arise for other ASOs? Second, is it specific to a brain with Huntington’s disease or would the same thing happen if you gave the drug to healthy controls or to people with premanifest HD? Dr. Schobel acknowledged of these questions as he addressed the topic of “class risks” and mentioned plans for post-hoc analyses stratifying by disease stage. Remember that nusinersen is a highly successful approved drug that — there is simply no doubt — is greatly improving outcomes for kids with SMA, some of whom have been on drug for several years now. This argues it cannot categorically the case that ASOs in the brain are not tolerated on chronic dosing. Still, it could hypothetically be that tominersen was one very rare bad apple and we just got unlucky, or it could be the case that it reflects a toxicological liability shared by some appreciable fraction of potential CNS ASOs. Meanwhile, in animal models of HD, there exists just one precedent (see [Southwell 2018] Figure S3) for an ASO being tolerated in healthy animals but not well tolerated in sick animals — but such an effect was not reported for tominersen.
implications
I wrote in my previous post that we still didn’t know what happened in GENERATION HD1 and it was too early to draw conclusions about any implications for our work in prion disease. That’s largely still true. If the unfavorable effect of tominersen is on-target, related to HTT lowering, then it has nothing to do with our program. If it’s off-target, then we look to the questions of whether it affects one ASO or many, and whether it’s disease stage-related (and if so, is it HD or other diseases too). Ionis has trials ongoing of multiple other CNS ASOs in symptomatic populations, including those targeting MAPT in PSP (NCT04539041) FUS in ALS (NCT04768972) and GFAP in Alexander disease (NCT04849741), plus most importantly, tofersen for SOD1 ALS, which will read out late this summer (NCT02623699). Thus, over time we will have progressively more data to help rule in or rule out any implications of the tominersen failure for CNS ASOs more broadly.
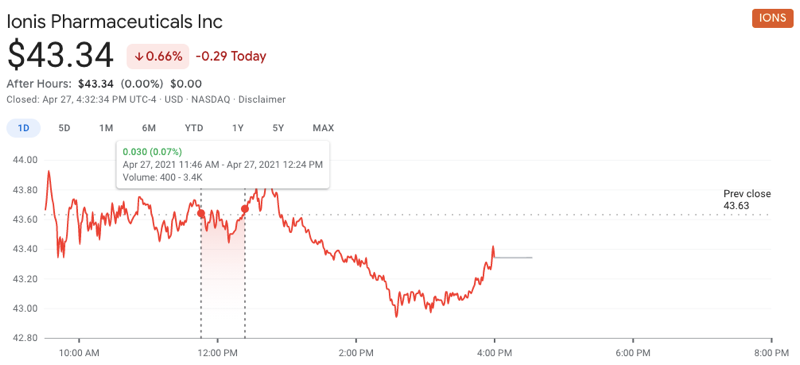
In the meantime, for what it’s worth, Wall Street seems not to have felt that the details announced yesterday had any implications for Ionis’s broader portfolio. After some technical difficulties, the talk got underway at 11:46a, and had transitioned to Q&A by 12:24p. Over this time, you can see that there was no appreciable movement in Ionis’s stock price, and indeed, the overall change for the whole day was inconsequential (note the tight y axis):
Another question on our minds is whether these results have any implication specifically for the use of ASOs in preventing, as opposed to treating, neurodegenerative disease. Here, it could push either way. On one hand, the fact that tominersen had a bad outcome could make regulators, companies, investigators, as well as potential trial participants, more cautious about the prospect of giving an ASO to still-healthy people. On the other hand, if evidence eventually emerges that the effect was disease stage-related, that could intensify interest in intervening at earlier stages, both in premanifest HD and potentially in other neurological disorders. A lot will probably depend on the upcoming analyses that Dr. Schobel mentioned, and on the outcomes of other neurological ASO trials.
Finally, another question is whether these results will have implications for the duration or size of future ASO trials. Here, I lean towards likely yes: the fact that unfavorable effects of tominersen emerged over 17 months of Phase III that were not observed in the 4 months of Phase I in a smaller number of participants suggests that for future ASOs, regulators might expect to see a longer drug exposure, possibly in a larger number of individuals, to rule out a similar effect before approval.
There is no doubt that for the HD community, tominersen’s failure is a huge setback. But I agree with Dr. Schobel’s assessment that there is a wealth of knowledge to be learned from this trial, particularly through the continued collection of outcome measures from participants even though dosing is stopped. So the HD patients who volunteered for this trial did not do so for nothing — rather, it was a courageous act that will no doubt move the field forward in the long run, and we should continue to hail them as heroes. And while it may be cold comfort for people suffering from HD now and wanting an effective treatment today, the CHDI meeting’s agenda is an impressive roundup of scientific progress being made across many domains, with many drug candidates in development. For those outside the HD community, such as ourselves, the tominersen results are an important development to be aware of, that are likely to come up again and again in the future, but it is still too early to draw any firm conclusions about what it means for us. In the meantime, we’re grateful that Ionis has recently reaffirmed its commitment to prion disease, and for our research, it’s full steam ahead.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.