Prevention and targeted therapies are not business as usual
In a new paper published today [Mortberg 2022], we analyze 20 years of clinical trial registrations for neurodegenerative diseases in order to map the landscape of drug development in these disorders. At a high level, the biggest take-home for us is just how much our approach — championing preventive intervention with a targeted therapy — is not business as usual. In this blog post I’ll unpack the motivation for the study, the different questions we asked, and what picture it paints.
Several years ago, Sonia and I started making a noise about the need for prevention in genetic prion disease. Early on, some people dismissed this as being impractical, saying that’s just not how trials are done, and regulators would never accept it. This launched us on a mission to engage early with regulators and think through what it would take to do a true primary prevention trial in prion disease. But years later, I still felt that a piece of data was missing: what is the way trials are done? What is business as usual? When I looked around for a quantitative analysis of the landscape of trials in neurodegenerative disease, I never found quite what I was looking for. So we set out to do it ourselves. In a project led by Meredith Mortberg from our lab, we queried ClinicalTrials.gov for trials in Alzheimer’s, Parkinson’s, Huntington’s, and FTD/ALS from 2000-2020, and we went deep on annotating and analyzing the characteristics of those trials. Here are a few of the most interesting things we learned.
First: while there are all kinds of trials out there, industry-sponsored drug trials appear to represent the lion’s share of credible “shots on goal”. In terms of sheer number of trials, there are actually more trials run by academics, government, or non-profits than by industry. And a substantial fraction of trials test medical devices, procedures, or behavioral interventions. On average, though, industry drug trials are larger/longer, such that they account for 61% of all patient-years. What’s more, industry drug trials are 2x more likely to run to completion and 3x more likely to be placebo-controlled. Whether any particular trial is statistically “well-powered” for its endpoint is of course a devilishly complicated question based on myriad assumptions and a ton of data we don’t have in this high-level retrospective analysis. So it’s impossible to say which specific trials in this dataset were or weren’t well-powered. But in the aggregate, if you want to transform the awful prognosis of neurodegenerative disease, you probably want statistically robust evidence that a specific intervention is better than no intervention, and in the aggregate, it looks like industry drug trials are a lot more likely to be able to deliver such evidence than any other kind of trial.

Above: dot plot cross-tabulation of clinical trials. The size of the dot represents the number of trials, and the color represents the “intensity” — the number of patient-years per trial. In terms of sheer count of trials (dot size), there are plenty of trials of behavioral interventions and of devices, plenty of trials with non-industry sponsors, plenty of trials in Parkinson’s. But in terms of patient-years (probably a better proxy for dollars spent), investment in trials is much more concentrated in industry-sponsored drug trials — especially Phase III in Alzheimer’s.
Second: trials have gradually shifted to focus on less-impaired participants, at the expense of being more picky. 57% of trials used some kind of cognitive or functional test as an eligibility criterion — usually MMSE in Alzheimer’s or Hoehn & Yahr in Parkinson’s. The score ranges that were eligible to enroll in a trial shifted towards the less impaired end of the spectrum over the 20 year period. In particular, the minimum level of impairment required to enroll dropped a bit, so that less-impaired people became eligible, but the maximum level of impairment permitted dropped even more, thus screening out too-impaired people. On the net, the size of the acceptable score range shrunk — a narrower window was eligible. That’s right, trials have gotten pickier, one reason why it’s so hard to enroll enough Alzheimer’s patients these days. For industry drug trials, we can see this same trend in the simple number of inclusion/exclusion criteria listed in the ClinicalTrials.gov entry — the average number of criteria listed nearly doubled from 2000 to 2020. This was especially true for Phase III trials but was significant even for Phase I and II.
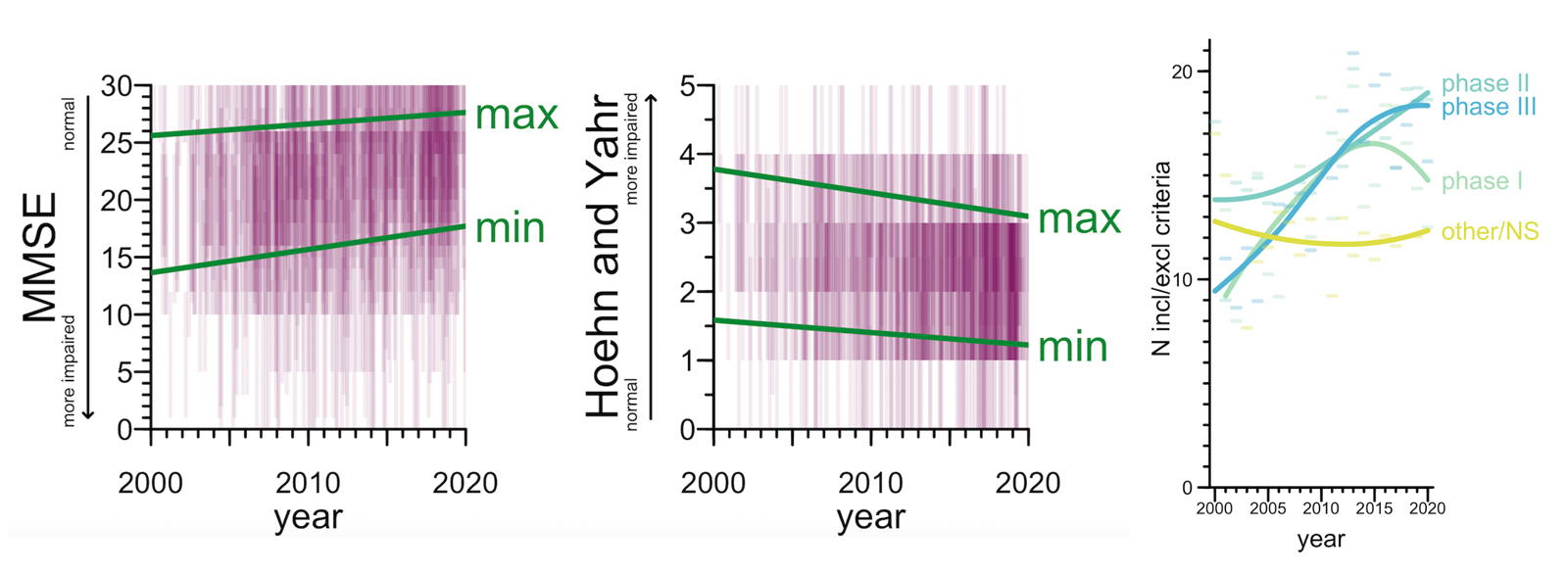
Above: (left) eligible score ranges on MMSE and Hoehn & Yahr. Semitransparent purple rectangles represent ranges in individual trials, green lines represent linear regressions on the min and max score. (right) Number of inclusion/exclusion criteria per trial. Dashes represent means per year, curves represent loess fits.
Third: prevention remains vanishingly rare. That shift towards slightly less-impaired participants that I just described mostly just reflects different cutoffs among already-symptomatic patients. The alternative — trials that actually go out and recruit patients who are not yet sick — has been and remains extremely rare. In all, just 2.7% of trials (89 of them) were open to pre-symptomatic participants. What’s more, those 2.7% of trials looked pretty different from the other 97.3% of trials. Preventive trials were less likely to be industry-sponsored, less likely to test a drug, and much more likely to test a behavioral intervention.
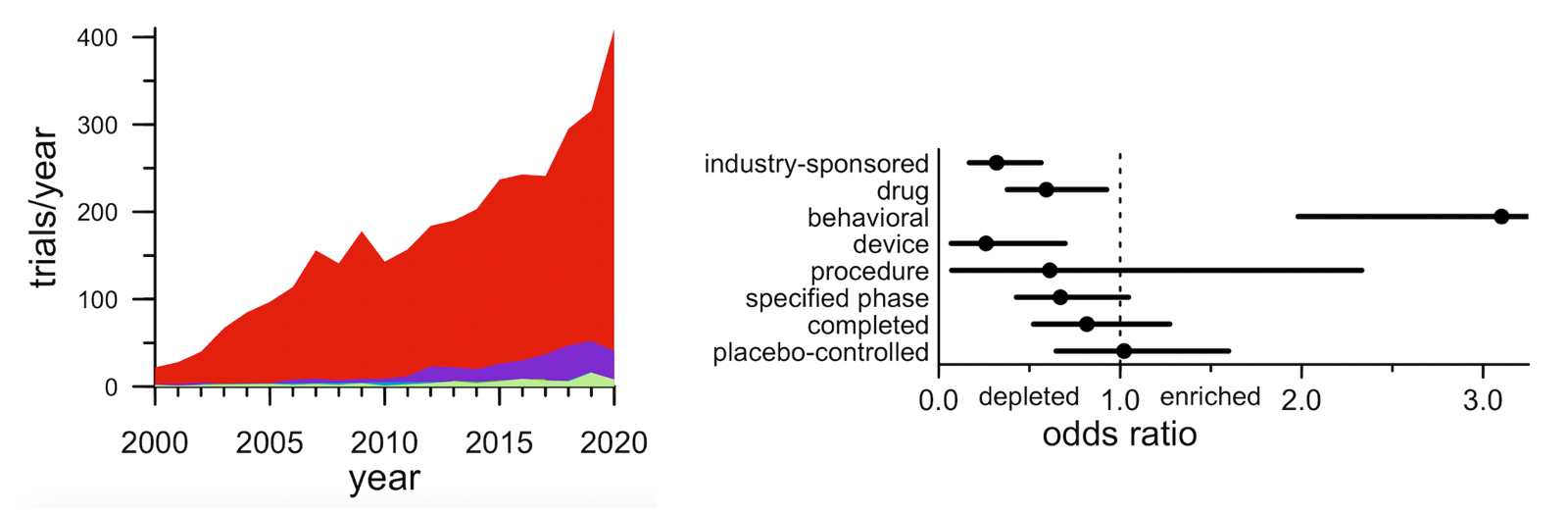
Above: (left) stacked area plot of number of trials by year by disease stage. The red is symptomatic patients with a diagnosis of AD, PD, HD, or FTD/ALS; the purple is “mild” stages such as mild cognitive impairment (MCI); the blue and green that you can barely see are different pre-symptomatic stages. (right) Forest plot showing how the characteristics of presymptomatic trials differ from symptomatic trials.
Fourth: few trials test a mechanism causally linked to disease through human genetics. A lot of what I blog about, and what we hear about in the biotech/pharma news, is trials aimed at core disease mechanisms: SOD1 in ALS, HTT in Huntington’s, Aβ in Alzheimer’s. Those are all cases where the drug goes after a target that we know from human genetics (and often many other sources of evidence too) is core to the disease process. I kind of thought that’s what most trials in neurodegeneration are focused on. Turns out it’s not. Trials that tested a therapeutic hypothesis supported by a human genetic association were just 8% of trials, or 16% of patient-years. And those numbers did not rise from 2000 to 2020, even though the number of known genetic associations grew. Viewed through one lens, this is partly because of lag: it took an average of 13 years for a hypothesis to get from genetic discovery to first clinical trial. Dissecting a mechanism, and then developing a drug around it, is slow work. But many of the Mendelian causes of neurodegenerative diseases were known before 2000, and yet never became the focus of more than a small minority of trials. So taking a different lens, we can also ask, what are all those other trials? The biggest category, about a third of patient-years, is trials of other novel agents, for most of which we could not identify any known mechanistic target. Maybe someone out there hypothesizes they’re going to be fundamentally disease modifying, but there aren’t even enough data to say what (if anything) the mechanism of action is, let alone how that mechanism might be relevant to disease. Another large bucket is repurposing trials — attempts to reposition a drug already approved for some other disease. A few of these may be rooted in some firm mechanistic hypothesis, but many are just attempts to manage symptoms better. For instance, the most-studied repurposed drug was botulinum toxin, which is used in Parkinson’s to manage involuntary movements and bladder contraction. The most surprising category of trials, to me, was trials of drugs that are already approved, for the same indication for which they are already approved. Donepezil was approved to manage symptoms of Alzheimer’s in 1996, and yet from 2000 to 2020 there were 56 additional trials of donepezil in Alzheimer’s. These trials examined all sorts of questions, such as the drug’s efficacy at different disease stages, its impact on certain outcomes such as brain imaging metrics, its interaction with genetic markers, and so on. All in all, for every one trial that led to approval of a drug, there were more than four trials after approval.
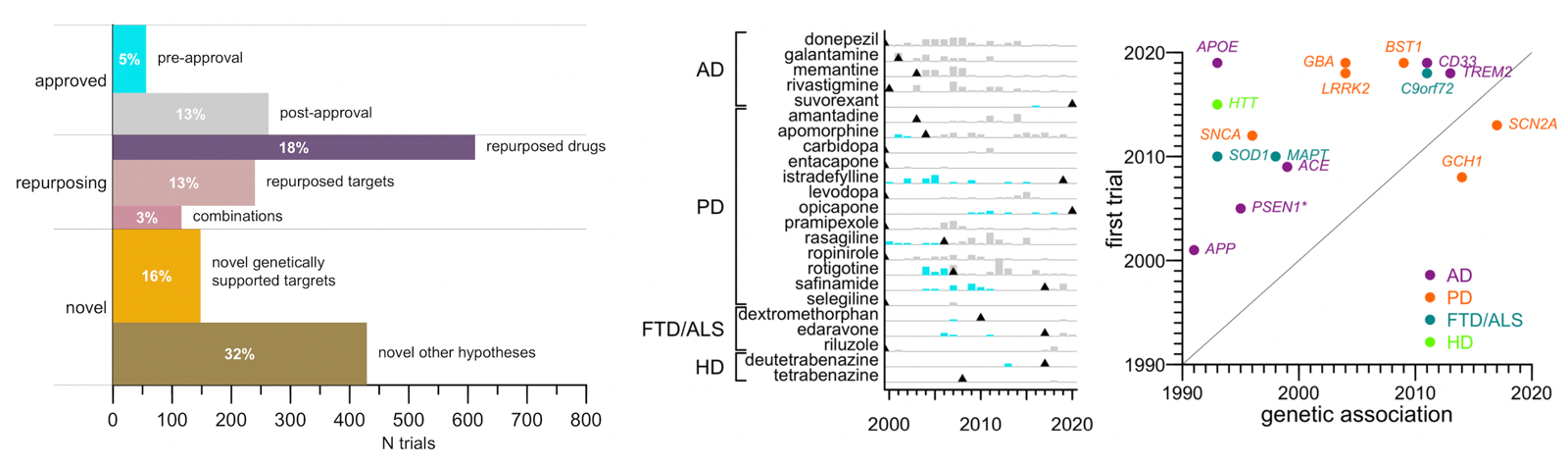
Above: (left) proportional area barplot of categories of clinical trials in neurodegeneration. Length represents number of trials while area (and indicated percentages) indicate share of all patient-years. (center) barplot of trials before (cyan) and after (gray) approval for neurodegenerative disease drugs. (right) scatterplot of time from genetic association to first trial of that mechanism.
For me, these findings were illuminating, and some were a bit depressing.
At some point in the past, without explicitly realizing it or articulating it, I held the following naive view: even if “cures” are rare, everyone sure wants a “cure”, and therefore, every clinical trial must represent a separate attempt to fundamentally transform the treatment of a disease. I now see that a large proportion of drug development effort is aimed at the margins — trying to slightly expand the label for donepezil even though everyone agrees donepezil will not suddenly become a cure. Of course, I am rooted in thinking about an ultra-rapid disease. Patients with these more slowly progressive disorders, where you have to live with symptoms for years and years, will appreciate the value of symptom-managing therapies in a way that I fail to.
I must have also believed this: that our newfound lens into the fundamental molecular drivers of disease, gained through genetics, must be refocusing effort in drug development. Why would we take shots in the dark now that the lights are on? But maybe from a sponsor’s standpoint, therapeutic hypotheses can get their appeal from different sources. Some drugs might be appealing because they have good scientific evidence behind the mechanism, but may in turn have the liability of being novel modalities or of having big open questions about long-term safety and pharmacology. Others might have a low probability of success or at least a low probability of really modifying disease, but be cheaper to test because they have already been in humans. So all kinds of trials get done, and for those of us hoping for a fundamental transformation in treatment, only a fraction of those are trials in which we can place much of our hope.
Then there’s the question of disease stage. Ask anyone at risk for dementia: do you want a drug to prevent it, or to slow it down once it strikes? I have yet to hear someone make the case for why a slowing of symptomatic course is what they want for their own brain, or the brain of their loved one. So why do the overwhelming majority of trials aim at slowing rather than preventing? The answer, of course, is a laundry list of practical considerations: the duration of follow-up required to demonstrate prevention, the biomarkers (or lack thereof) available as preventive endpoints, the safety risks of taking a novel drug into otherwise-healthy people. Among the few scientists and clinicians I have really talked to at length about this, it appears that most do aspire to prevention as the ultimate goal. But they feel it is impractical as an initial clinical strategy for most diseases or most drugs. So instead they invest their hope in the idea that a drug will prove to have enough benefit at a symptomatic stage to get it approved, and hope in the long run be able to demonstrate a much larger benefit in a preventive setting. A concern, then, would be if the molecular mechanisms of disease initiation are distinct from those of disease progression. And in some human genomic studies, what we see is that there is only a limited overlap between the genetic risk factors that contribute to initiation and those that contribute to progression. If so, does 97.3% of trials being in sick patients mean a lot of opportunity to miss a benefit that would have been achievable in pre-symptomatic patients? We recently learned that Aβ antibody lecanemab slowed cognitive decline by 27% over 18 months in an Alzheimer’s trial. Despite the modest effect size, that is fantastic news, because it validates that this one target, at least, is in fact relevant across disease stages. Still, only a fraction of trials are actually testing a hypothesis with good mechanistic roots, and the vast majority of that investment has been on Aβ itself. It took 100, (yes, 100) trials of Aβ-targeting drugs to yield this 1 success. Can we count on getting this many shots on goal for other targets like C9orf72 and LRRK2?
This research was really just an attempt to describe what’s been done in neurodegenerative disease trials, and the above interpretations are just my opinion. People will take away from the study what they want to, and certainly this paper does not provide any remedy for the things that are challenging about preventive trials. But in the ongoing debate about where and how we should invest our resources in the fight against neurodegeneration, I hope this all can provide one point of reference and some basis for reflection.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.