Finding all the polyQ genes in the human genome
Recently I had reason to want a list of all the polyglutamine (polyQ) genes in the human genome. This seems like it should be a really trivial thing to conjure up, yet I found it surprisingly difficult. Here’s what did and didn’t work.
tl;dr: What did work: I downloaded a FASTA file of the human proteome from uniprot, containing the amino acid sequences for ~87K proteins. It looks like this:
$ head HUMAN.fasta >sp|A0A183|LCE6A_HUMAN Late cornified envelope protein 6A OS=Homo sapiens GN=LCE6A PE=2 SV=1 MSQQKQQSWKPPNVPKCSPPQRSNPCLAPYSTPCGAPHSEGCHSSSQRPEVQKPRRARQK LRCLSRGTTYHCKEEECEGD >sp|A0A5B9|TRBC2_HUMAN T-cell receptor beta-2 chain C region OS=Homo sapiens GN=TRBC2 PE=1 SV=1 DLKNVFPPEVAVFEPSEAEISHTQKATLVCLATGFYPDHVELSWWVNGKEVHSGVSTDPQ PLKEQPALNDSRYCLSSRLRVSATFWQNPRNHFRCQVQFYGLSENDEWTQDRAKPVTQIV SAEAWGRADCGFTSESYQQGVLSATILYEILLGKATLYAVLVSALVLMAMVKRKDSRG >sp|A0AUZ9|KAL1L_HUMAN KAT8 regulatory NSL complex subunit 1-like protein OS=Homo sapiens GN=KANSL1L PE=1 SV=2 MTPALREATAKGISFSSLPSTMESDKMLYMESPRTVDEKLKGDTFSQMLGFPTPEPTLNT NFVNLKHFGSPQSSKHYQTVFLMRSNSTLNKHNENYKQKKLGEPSCNKLKNILYNGSNIQ
And since I’m always thinking relationally, I wrote a one-minute Python script to convert the FASTA into a tab-separated format so that the amino acid sequence for each gene would be on the same line as the spid (uniprot’s unique identifier) and gene symbol (if any):
import sys
i = sys.stdin
o = sys.stdout
prot = ""
for line in i.readlines():
if line[0] == ">": # start of a new FASTA header
# the previous protein is now done, write it out and start over.
if prot != "":
o.write("\t".join([spid,genesymbol,fulldesc,prot])+"\n")
prot = ""
spid = line.strip().split()[0].split("|")[1] # parse spid from line
# parse gene symbol if any
if ("GN=" in line):
genesymbol = line.strip().split("GN=")[1].split(" ")[0]
else:
genesymbol = ""
fulldesc = line.strip() # and just grab the full description of the protein
else:
prot += line.strip() # for non-header lines, concatenate the amino acid sequences
o.write("\t".join([spid,genesymbol,fulldesc,prot])+"\n")
Here’s how use that script:
cat HUMAN.fasta | python uniprot-to-tab.py > uniprot.tab
Now say you want to find all proteins with at least 8 consecutive glutamines. If you’re Unix-minded you can find the polyQ genes like so:
grep QQQQQQQQ uniprot.tab
Or if you’re SQL-minded you can read it into a table:
drop table if exists uniprot_human;
create table uniprot_human (
upid serial primary key,
spid varchar,
genesymbol varchar,
fulldesc varchar,
prot varchar
);
copy uniprot_human (spid,genesymbol,fulldesc,prot) from 'c:/sci/019enrep/data/uniprot/uniprot.tab' with null as '';
And then find the gene symbols like so:
select distinct genesymbol from uniprot_human where prot like '%QQQQQQQQ%' order by 1;
Answer: there are 97 of them. The advantage of having it in a SQL database is, say you want to know what transcripts are involved rather than what proteins, you can join the uniprot table to UCSC’s kgXref table on spid. kgXref can be found in the UCSC table browser as follows:

Though this is all easy enough, it may not be a final answer depending on what your ultimate interest is. kgXref will let you map from protein to RefSeq mRNA, but say you want to know the genomic coordinates of the DNA that codes for the polyQ repeat – then you’ve got a messy mapping process ahead of you [see this biostars discussion].
Aside #1: some things that didn’t work, at least not readily. UCSC table browser also has a list of all the simple repeats at least 24b in length:

Which is a nice resource. But in order to figure out which repeats are protein-coding, you need to join it to something like the RefSeq Genes table:

Which is quite non-relational: exon starts and ends are given in a comma-separated list within a field, and exons include 5′UTR and 3′UTR and non-coding transcripts, so you need to only grab the pieces of exons that are inside the coding sequence (cdsStart:cdsEnd). And according to the annotation, the coding exons sometimes start mid-amino acid (in the below example, a glycine is supposedly coded-for by just two DNA bases):
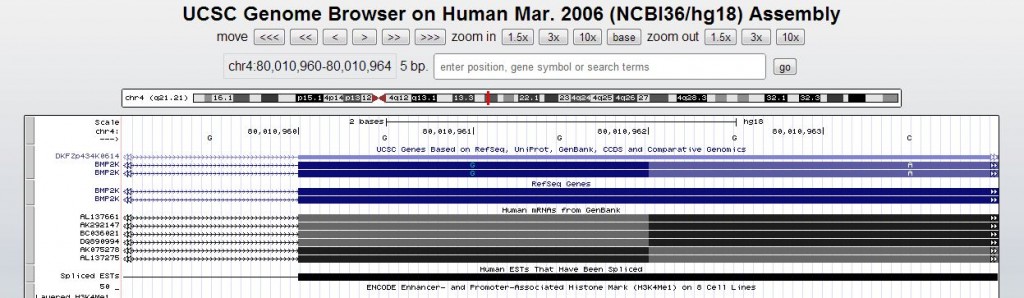
Which means that you can’t just trivially use modular division to figure out what exonic frame the repeat falls in. The RefSeq Genes table has an exon frame field, but it appears those are given relative to the exon starts rather than in absolute terms. It’s all fairly complicated and I could find no documentation on how to interpret these table fields, so I eventually gave up and went the proteome route as described above.
Aside #2: the just Google it approach. Before I tried any of this, the first thing I did was Google a few search terms like “list of polyq genes” and “list of polyglutamine proteins”. That didn’t turn anything up, which is why I embarked on figuring it out myself. Then after I had already finished I was Googling something else and stumbled upon Robertson’s polyQ database [Robertson 2011]. Supposedly that has the answers I was looking for anyway, along with a host of other annotations. But actually I don’t see any way to just get a list of all the proteins. The help page says “All PolyQ-containing proteins will be displayed when the ‘All Proteins’ box is clicked”, but in fact nothing at all is displayed when I click the All Proteins box. So maybe it was worth going to all this trouble after all.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.