Biochemistry 10: lipid metabolism
These are notes from lecture 10 of Harvard Extension’s biochemistry class.
intake and distribution of fats
Fat can be consumed directly in the diet or derived (by the liver) from excess dietary carbohydrates. Once stored, it can be re-mobilized from adipose tissue. Typical humans (in developed countries??) get ~40% of their calories from dietary fat.
Triacylglycerols (TAGs) are broken down by lipases. Pancreatic lipases in the intestinal lumen help to absorb fatty acids from the diet into the intestine. Lipoprotein lipases in the capillary walls help absorb fatty acid from chylomicrons and VLDLs into target tissues. Hormone-sensitive lipases inside cells break down fat stores in adipose tissue.
Bile salts are various derivatives of cholesterol with different R2 groups at the end of the chain. The bile salt acts as a detergent, breaking apart large dietary globules of fat to yield smaller micelles, which are more accessible to lipases than the large globules. Intestinal lipases then convert TAGs into mono- and di-acylglycerols, free fatty acids, and glycerol. Now the TAGs need to somehow travel through the blood to be of use as energy to other organs. This is accomplished by packaging them into lipoproteins.
A lipoprotein is a ball with a single-layered phospholipid + cholesterol surface, filled with triacylglycerols and cholesterol esters (cholesterol with a fatty acid attached). Apolipoproteins are embedded in the surface.
| class | abbreviation | density | protein | lipid content |
|---|---|---|---|---|
| chylomicrons | CM | lowest | lowest | highest |
| very low density lipoproteins | VLDL | .. | .. | .. |
| intermediate density lipoproteins* | IDL | .. | .. | .. |
| low density lipoproteins | LDL | .. | .. | .. |
| high density lipoproteins | HDL | highest | highest | lowest |
*Yes, this is not a mistake; IDL is really in between VLDL and LDL, not between LDL and HDL.
Chylomicrons are an exogenous pathway of lipid metabolism because they are where dietary fats go directly. Chylomicrons appear in the blood about 2 hours after a meal and disappear from the blood about 16 hours after a meal (having been taken up by the liver). They are large and concentrated enough to visibly cloud your plasma.
VLDLs are an endogenous pathway because that’s where fats synthesized de novo in the liver or derived from adipose tissue go. CMs and VLDLs are distinguished by which APOB is on their surface.
HDL serves as a reservoir for different apolipoproteins and can exchange them with other lipoproteins.
Once in tissue (capillaries), lipoprotein lipases hydrolyze TAGs into fatty acids and glycerol. In muscle, fatty acids are oxidized for energy; in adipose tissue, they are re-esterized as TAGs for storage. Lipoprotein lipases bind CMs and VLDLs by interacting with APOC2.
As VLDLs are metabolized they lose lipid content, becoming more dense. They exchange APOC2 for APOE and become IDL particles. APOE is a high-affinity ligand for receptor-mediated clearance. About 50% of IDL is taken up into the liver, and 50% will go on to lose APOE and become LDL. (All these particles have APOB100, and they retain this when they become LDL.)
LDL is taken up into the liver and other tissue by receptor-mediated endocytosis. Tissue receptors specifically bind APOB100 and LDL goes into the endosomal-lysosomal system where the cholesterol esters are hydrolyzed, and free cholesterol and fatty acid are released into the cytosol. The LDL receptor is recycled to the surface, and the free cholesterol goes into the ER where it inhibits HMG-CoA reductase, downregulates synthesis of LDL receptors and upregulates ACAT. [reviewed in Cohen & Armstrong].
Acyl-CoA-cholesterol acyl transferase (ACAT) is a liver enzyme which catalyzes the transfer of fatty acids to cholesterol to yield cholesterol esters. This is useful because the cholesterol esters are easier to package into lipoproteins than straight cholesterol.
LDLs have been dubbed “bad cholesterol” due to their association with atherosclerosis, in which fatty plaques build up on the inside of arteries. Why are LDLs are risk but not VLDLs, even though both transport cholesterol? Because LDL has only APOB100, which has only low affinity for the LDL receptor, so it stays in the plasma longer (2-4 days), giving it more time to stick to the walls. VLDLs have APOC2 which is much higher affinity, so they’re cleared more quickly.
HDL functions as a cholesterol scavenger, extracting excess cholesterol from cell surfaces and transporting it back to the liver (“reverse cholesterol transport”). Its concentration is inversely associated with cardiovascular disease (CVD), and thus it has been dubbed “good cholesterol.” However there are subtypes of HDLs. HDLs without APOC3 are probably good, but HDLs with APOC3 (~13% of HDL particles) are positively correlated with CVD. The level of APOC3 appears to be in part genetically determined.
utilization of fats for energy
Stored TAGs in adipose tissue are used as a source of energy whenever fuel stores are low. Glucagon and epinephrine trigger hormone-sensitive lipase to release fatty acids from fat stores in adipose tissue. The fatty acids are then transported to tissue (skeletal muscle and heart) for oxidation and energy production.
Hormone-sensitive lipase cleaves triacylglycerol into glycerol and three fatty acyl groups. Glycerol is converted to glyceraldehyde 4 phosphate which enters glycolysis as an intermediate. That pathway provides 5% of the energy released by TAG metabolism. The other 95% comes from the beta-oxidation of the three fatty acyl groups.
On the cytosolic side of the outer mitochondrial membrane, acyl-CoA synthetase uses ATP to add a CoA to fatty acid, yielding fatty acyl-CoA. In fact, it double-spends the ATP, converting it to AMP and releasing two Pi. Fatty acyl-CoA is then transported into the mitochondrial matrix by the carnitine shuttle. Once inside the matrix it undergoes beta oxidation.
Each round of beta oxidation has the net effect of removing two carbons from the fatty acid chain. It also releases one acetyl-CoA, and replaces it on the end of the fatty acid chain. Each round of beta oxidation directly produces 1 FADH2 and 1 NADH to feed into the electron transport chain, and yields one acetyl-CoA which can then be oxidized in the citric acid cycle to yield additional 3 NADH, 1 FADH2 and 1 GTP. So from one round of beta oxidation, you get ~14 ATP.
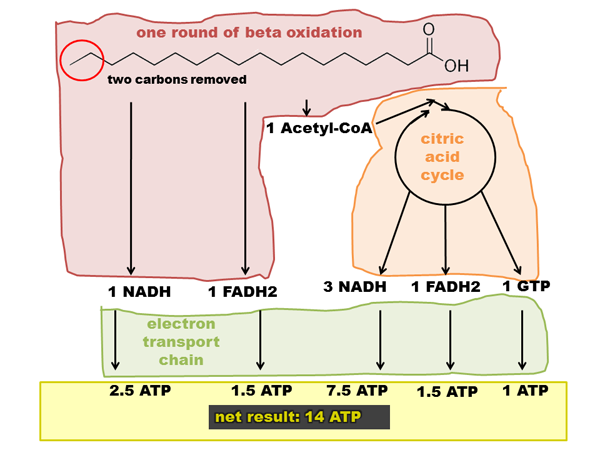
So if you start with a 16-carbon fatty acid like palmitate (converted to pamitoyl CoA), you can run 7 rounds plus then be left with 1 acetyl-CoA (which contains the final 2 carbons). So the metabolism of one palmitate yields 8 acetyl-CoA, 7 FADH2 and 7 NADH for a total of 108 ATP, minus 2 ATP spent for activation. Net yield of 106 ATP per 16-carbon fatty acid.
That was for even chain fatty acids. With odd chain fatty acids you get propionyl-CoA and eventually yield succinyl-CoA which enters the citric acid cycle. Thus, odd-chain fatty acids feed intermediates into the CAC, increasing CAC activity.
Fatty acid oxidation is regulated by hormonal regulation of hormone-sensitive lipase. Glucagon and epinephrine increase cAMP, which leads to a phosphorylation cascade which increases hormone-sensitive lipase activity, increasing free fatty acids and thus increasing beta-oxidation. There is also transcriptional regulation of fatty acid oxidation enzymes, mediated by the PPAR family of nuclear receptors. More on that in the next lecture. Finally, fatty acid oxidation is also regulated by malonyl CoA.
Comparison of fatty acid degradation and synthesis:
| degradation | synthesis | |
|---|---|---|
| tissue location | most tissues; especially high in liver, muscle and adipose | mostly liver and a little bit adipose |
| subcellular location | mitochondrial matrix | cytosol |
| electron situation | NAD+ is electron acceptor | NADPH is electron donor |
| energetics | 2 ATP used for activation of acyl group, 108 reaped for net 106 ATP | 1 ATP utilized for every 2 carbons incorporated (but obviously the molecules being incorporated are already high-energy) |
synthesis of fatty acids
Acetyl-CoA is diverted from the citric acid cycle towards fatty acid synthesis when high-energy molecules (ATP, NADH) are abundant. Acetyl-CoA produced in mitochonrdria has to be transported to the cytosol via the tricarboxylate transport system for fatty acid synthesis.
The first step in the synthesis pathway is the rate controlling step. It’s acetyl CoA carboxylase (ACC) adding a carbon from bicarbonate to yield malonyl-CoA. Then, fatty acid synthase (FAS) is a multifunctional protein, a single polypeptide chain but with multiple active sites each doing different things. Each domain catalyzes one reaction, and there’s an “ACP arm” that swings around carrying the intermediates from one site to another.
Synthesizing 1 palmitate takes a lot of energy: 1 acetyl CoA directly and another 7 acetyl CoA for the production of malonyl-CoA, 7 ATP used in that acetyl CoA – malonyl CoA conversion, and 14 NADPH for reducing power in two different active sites of FAS, and 1 ATP to transport the acetyl-CoA from the mitochondrial matrix into the cytosol.
From palmitate, we have elongases which add additional carbons and desaturases which introduce double bonds. Beyond oleate (18:1Δ9), mammals cannot go any further – we can’t introduce double bonds beyond C9, yet we do need some of those. These are essential fatty acids which can be synthesized only by plants and must be obtained in our diet. They are essential for instance for making arachidonate.
Products of carbohydrate and amino acid catabolism are directed towards fatty acid synthesis when fuel is abundant. Synthesis is regulated at the ACC step. Citrate (a sign of abundant energy) allosterically stimulates ACC, and palmitoyl CoA (a sign of excess fatty acids) inhibits it.
Malonyl-CoA inhibits carnitine acyltransferase to prevent fatty acids from being taken into the mitochondrial matrix to be beta oxidized at times when fatty acids are being synthesized, thus preventing a futile cycle.
Fatty acids that are ingested or synthesized have one of two fates: incorporation into TAGs for energy storage, or incorporation into phospholipids for membranes. TAGs are synthesized using dihydroxyacetone (a glycolysis intermediate). Insulin stimulates the conversion of acetyl CoA (thus ultimately dietary carbohydrates and amino acids) into fatty acids.
Cholesterol is used for three essential purposes: in cell membranes, as a precursor for hormones, and as a precursor for bile acids. In cholesterol synthesis, 2 acetyl-CoA are converted into acetoacetyl-CoA and then into HMG-CoA. HMG-CoA reductase converts this into mevalonate. Mevalonate is diphosphorylated from ATP to yield an activated isoprene which can be made into cholesterol. HMG-CoA reductase is the rate determining step of cholesterol synthesis and a major control point. It is stimulated by insulin, inhibited by glucagon, and transcriptionally regulated by intracellular cholesterol. (More cholesterol means downregulation of HMG-CoA reductase).
SCAP is a sterol sensor which complexes with sterol regulatory element (SRE)-binding proteins (SREBPs). When cholesterol gets low, SCAP allows SREBP to proceed to the Golgi where it is cleaved by two different proteases to liberate a cytosolic domain which binds to the SREs in promoter regions and transactivates HMG-CoA reductase (HMGCR).
Statins are competitive inhibitors of HMG-CoA reductase. They not only reduce endogenous cholesterol synthesis, they also increase LDL receptors, thus reducing blood levels of cholesterol.
In familial hypercholesterolemia (FH), patients are deficient in LDL receptors (LDLR gene; there are a variety of different mutations) which leads to accumulation of blood cholesterol combined with intracellular cholesterol synthesis and ultimately atherosclerosis. Homozygotes can have heart attacks at age 5. Heterozygotes develop cardiovascular problems in their 30s.
Fatty acid oxidation in the liver yields acetyl-CoA which goes into the citric acid cycle but also produces ketone bodies. Ketone bodies can be exhaled as acetone or transported to extrahepatic tissue as an energy source in the form of acetoacetate and beta-hydroxybutyrate. During starvation, ketone bodies are an important source of fuel, especially in the brain. However, if ketone body production in the liver is in excess of the rate at which extrahepatic tissue can metabolize it, then blood pH drops, leading to coma and death. “Ketogenic diets” are high in fat, moderate in protein and low in carbohydrates. This forces your body to produce ketone bodies.
aside
Above it was stated that, besides reducing cholesterol, statins upregulate LDLR. That’s interesting because statins prolong life in prion-infected mice and we don’t know why – some studies have found no change in cholesterol. Overexpression of LDLR in the brain promotes amyloid beta clearance [Kim 2009].

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.