The overdrive hypothesis: is alpha cleavage key to keeping PrP in check?
Last year, I blogged a general introduction to alpha and beta cleavage, two events in the proteolytic processing of PrP. While the paradigm introduced in that post remains broadly correct, some refinements may be in order following recent work from Glenn Millhauser’s group [McDonald 2014]. While there are implications for beta cleavage and shedding as well, to me the most interesting part was the new view of alpha cleavage. I’ve attempted to diagram a subset of the findings below.
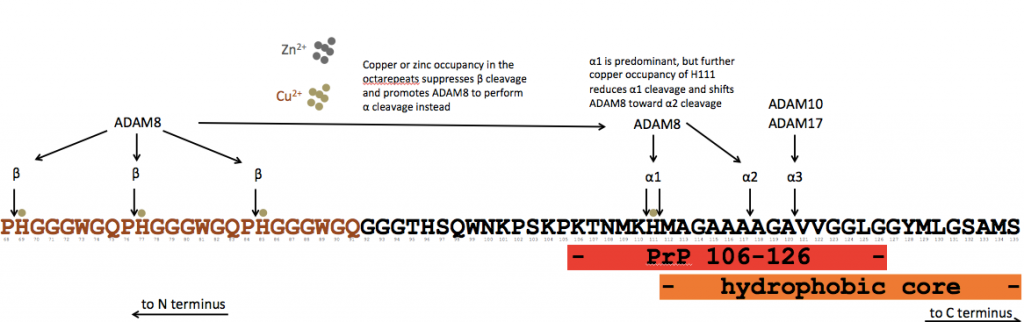
I’ve used human numbering and sequence, though the study used recombinant MoPrP. The study was done entirely in vitro with purified ingredients, and the relative prevalence of the new cleavage events in vivo remains to be confirmed. The study finds that there are actually three distinct types of alpha cleavage – α1, which we already knew about, plus α2 and α3 which occur a few residues further to the C terminus. In general, ADAM8 can perform β or α cleavage, but it seems to not like to cut at sites where a metal ion is bound, so introduction of a bit of copper, which occupies the octarepeats with higher affinity, reduces beta cleavage and shifts ADAM8 toward α cleavage. (Zinc does the same). Introduction of even more copper leads to occupancy of H111, which shifts the balance away from α1 and towards α2 cleavage. In recombinant MoPrP with the equivalent of the D178N or E200K mutations, the ability of copper to stimulate α2 cleavage is largely abolished. That’s interesting because other recent work from Millhauser’s group suggests that these mutants, which increase charge on a C-terminal face of the protein, reduce the inclination of PrP’s N terminus to interact with that face [Spevacek 2013].
If all that sounds like a lot of specifics, the big picture here is that this paints a picture whereby PrP proteolysis is regulated by the presence of metal ions. Alpha cleavage, mind you, is conserved at least out to chickens [Harris 1993], and seems to be required to prevent the peripheral demyelination phenotype of PrP knockout mice [Bremer 2010], so it must be intimately related to a native function of PrP.
In a new review in Prion, Millhauser argues that alpha cleavage might be a general mechanism for keeping PrP’s activity in check, and speculates that the neurotoxicity of PrPSc might arise from an uncontrolled exaggeration of such activity when alpha cleavage is impaired [McDonald & Millhauser 2014].
What “activity,” you ask? PrP has been shown to interact with a number of different glutamate receptors including AMPA [Watt 2013], NMDA [You 2012] and mGluR5 [Um 2013], so Millhauser uses as one specific example the concept [Watt 2013] that PrP helps load zinc into the AMPA receptor for uptake into cells. When zinc is present, PrP’s N terminus interacts with AMPA and the octarepeats provide zinc for import into the cytosol; but zinc also changes PrP’s tertiary structure in a way that makes it more accessible to alpha cleavage [Spevacek 2013], so that ADAM8 begins to cleave PrP, preventing the cell from being flooded with too much zinc.
If true, whether of AMPA and/or other interacting partners of PrP, this sort of story would elegantly tie together a few different observations. As Millhauser points out, the interstitital deletion mutants of PrP that cause spontaneous cerebellar degeneration are, without exception, those which abolish all of the alpha cleavage sites while leaving the N terminus intact. The “partial agonist” Δ114-121, which isn’t toxic on its own but also isn’t as good as PrPC at rescuing the toxicity of other mutants, lacks two of three alpha cleavage sites. In PrPSc, residues beyond 90 are often sequestered into proteinase K-resistant aggregates (and presumably first into lower-order oligomers), plausibly occluding the alpha cleavage sites from ADAM8′s reach.
Millhauser’s theory also seems to be reasonably compatible with other observations and other theories of PrP neurotoxicity. It certainly meshes with toxicity being mediated by PrP’s N terminal flexible tail [Sonati 2013], and the theory of an undampened interaction with AMPA or other channels is consistent with the spontaneous transmembrane currents observed in ΔCR cells [Solomon 2010]. And the theory leads to some testable hypotheses. If loss of alpha cleavage is responsible for the phenotype of ΔCR and other interstitial deletion mutants, then a shorter deletion, Δ110-121 in human sequence, which barely encompasses the three alpha cleavage sites, should be sufficient to recapitulate the ΔCR phenotype. Suppression of alpha cleavage by combining missense mutations – H111Y, V121D, and conversion of one or two alanines to aspartate in the palindromic sequnence in PrP should accomplish the same.
Granted, even if alpha cleavage is a key player here, that still doesn’t complete the story – the toxicity of globular domain ligands such as POM1 demands that some conformational change in PrPC is also required to induce the toxic “overdrive” [Sonati 2013]. There is a lot we don’t understand. And because Δ23-88 mice – lacking almost all of the N terminus of PrP – can still get fatal prion disease [Supattapone 2001], N terminal toxicity in all its forms simply cannot be the sole mechanism of neurotoxicity in prion disease – but that’s a topic for another post.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.