Genetics 05: 'Suppressor analysis in yeast'
These are my notes from lecture 05 of Harvard’s Genetics 201 course, delivered by Fred Winston on September 15, 2014.
Whole genome sequencing to identify mutants
Last time we discussed cloning by complementation. The other way to identify a mutant gene is by sequencing the whole genome of the mutant and the wild-type “littermates”. If you get 2:2 segregation you believe one mutation causes the phenotype, but because you mutagenized the genome, there are probably other mutations present as well. Suppose you find 100 variants unique to the mutant. To identify the 1 that is causal, people do pooled linkage analysis [Birkeland 2010]. We cross haploid cis1 by haploid wild-type CIS1, and we name our 100 mutant genotypes a,b,c,d,… and the wild-type alleles A,B,C,D,… Sporulate the diploids that result from your cross, dissect tetrads and identify the CisS and CisR progeny. Pool about 50 of the haploid CisR progeny and separately pool about 50 of the haploid CisS progeny. Sequence the pooled DNA. The causal mutation should have an allelic balance of 100% in the CisR library and 0% in the CisS library;. Unlinked non-causal mutations should center on 50/50 in both libraries. You might have a few linked non-causal mutations which hopefully will not quite segregate as perfectly as the causal mutation.
This is one of those situations where it is useful that yeast have a haploid life stage.
2-step gene replacement
5-fluoro-orotic acid (5FOA) has the following properties:
| genotype | phenotype |
|---|---|
| URA3 | Ura+, 5FOAS |
| ura3Δ | Ura-, 5FOAR |
5FOA is useful because you can do both positive and negative selection for 5FOA resistance because it is antithetical to the Ura+ phenotype.
Now, say we want to replace the CIS1 wild-type gene with the cis1 mutant. Suppose cis1 is caused by a A:T → G:C base pair change. Obtain a ura3Δ yeast strain, which has a CIS1 wild-type allele. Additionally obtain a URA3 plasmid. Amplify the URA3 gene from the plasmid using primers homologous to CIS1 flanking sequence. Introduce the amplified DNA into haploid cells, and this DNA will homologously recombine into the CIS1 locus. Select for transformants that have a Ura+ phenotype, and now you’ll have cells with ura3Δ in the original Ura locus, and URA3 in the CIS1 locus. Now transform these yeast with a 100 bp oligo containing the single base pair substitution found in the cis1 mutant. Now select for Ura- transformants, and you’ll get only those individuals where the synthesized cis1 mutant has recombined into the CIS1 locus, displacing the URA3 transgene. Now you have basically a cis1 knock-in. If this individual has the CisR phenotype, then that is your strongest evidence yet that the A:T → G:C variant you discovered in whole genome sequencing is indeed causal.
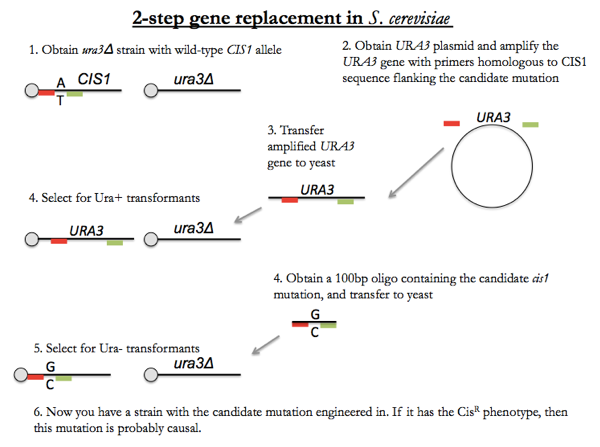
This technique is also used when you are using yeast as a system for functional studies of mutations found in humans. A candidate causal mutation in a human disease can be knocked into yeast using this method. This is also used when you perform yeast mutagenesis and you find there are multiple candidate causal mutations tightly linked in the same gene - you can do this method one by one for each mutation to figure out if one is sufficient for the phenotype.
Genome-wide approaches
A number of new technologies are useful for determining the mechanism by which a mutation affects function, or the native function of a wild-type gene.
RNA-seq
As one example, suppose we hypothesize that CIS1 affects transcription of other genes. We might perform mRNA-seq on wild-type, cis1 and cis1Δ strains. A typical mRNA-seq protocol involves the following steps:
- Select polyA RNA
- Make cDNAs
- Shear into fragments
- Ligate adapters and PCR amplify
- Perform Illumina sequencing
- Calculate transcripts per million or FPKMs or something like that for all genes
One might do this to simply compare wild-type to mutant in a baseline state, or one might look at different developmental stages such as meiosis, or might collect RNA after some environmental change such as oxidative stress. In S. cerevisiae, 30 million reads is generally considered sufficient for RNA-seq.
If you were to find that a number of genes were indeed differentially expressed in wild-type versus mutant, you might then ask whether the differentially expressed genes share a common regulatory sequence. Or one might ask whether the list of genes differentially expressed has a non-coincidental overlap with some other published gene list.
ChIP-seq
If we hypothesize that the Cis1 wild-type protein’s native function is to bind DNA at certain chromatin marks, we might perform ChIP-seq.
- Grow wild-type cells.
- Apply formaldehyde (CH2O) to cross-link amino groups separated by 2Å over a period of 30 minutes. Where proteins are bound to DNA, this will covalently attach them.
- Purify chromatin and sonicate the DNA to fragment it.
- Immunoprecipitate Cis1, using antibodies against Cis1 (or maybe you need an epitope-tagged Cis1)
- Reverse the cross-linking by heating to 65°C
- Sequence the DNA that co-IPed with Cis1
Tetrad example
In class we did a cross of trp1 × pan1. The results were 15 P, 13 N, and 2 T. P ≈ N, so the two genes are not linked to each other. The ratio is 1:1:<4, indicating that both markers are tightly linked to their centromeres.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.