Genetics 23: 'Genetics in families and the analysis of Mendelian traits'
These are my notes from lecture 23 of Harvard’s Genetics 201 course, delivered by Steve McCarroll on November 14, 2014.
Recombination and segment sharing in families
In humans there are on average 1-2 recombination events per chromosome per generation, for a total of about 30 across the whole genome. Our genomes are therefore mosaics of long segments that we inherit from recent ancestors.
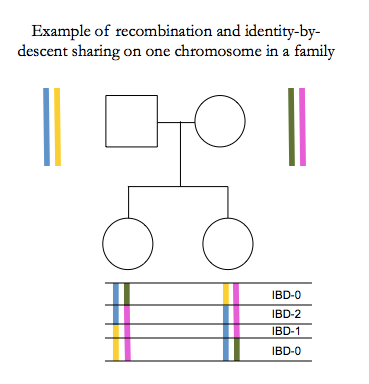
Total IBD between siblings is normally distributed 50% ± 4% [Visscher 2006].
Graham Coop has some great blog posts about this and related stuff - for instance this and this.
Monogenic (“Mendelian”) inheritance
Monogenic, oligogenic and polygenic traits are controlled by genetic variation at one, several or many loci respectively.
A more detailed key is available here.
{kind=link}
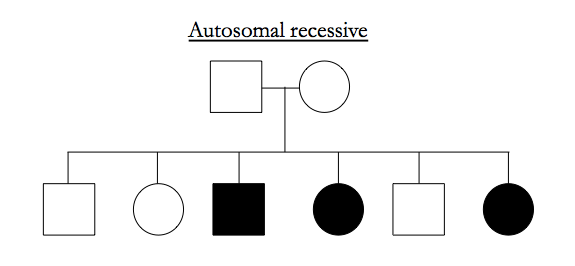
Here are examples of pedigrees suggestive of particular inheritance modes:
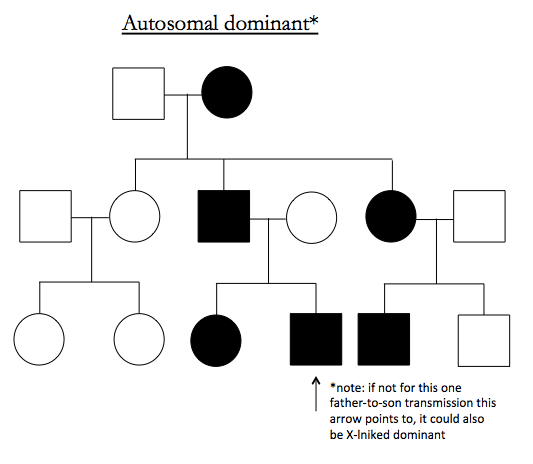
Note that X-linked dominant looks just like autosomal recessive except for an absence of father-to-son transmissions.
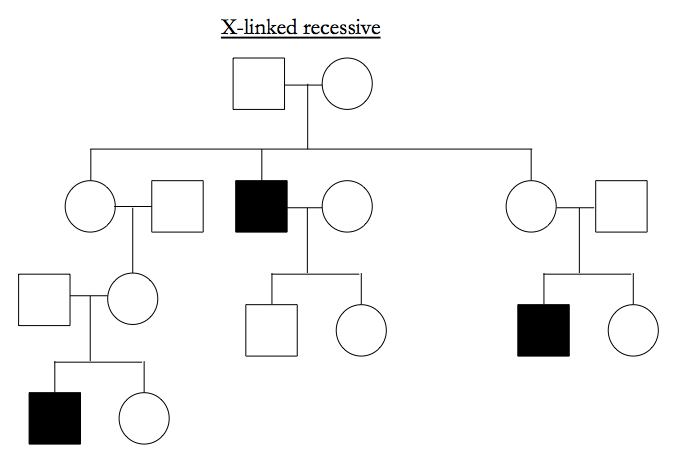
X-linked recessive typically affects males, the male has unaffected parents, and all the affected males in a pedigree share ancestry via mothers rather than fathers.
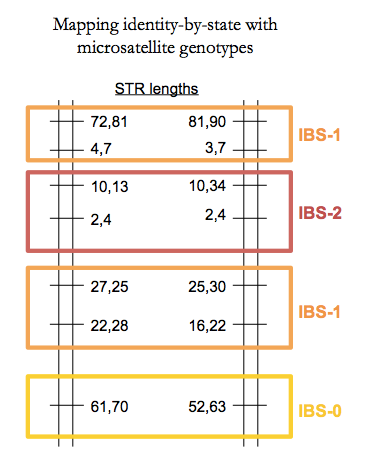
Recessive traits have to occupy parts of the genome that are IBD-2 among affected siblings.
Dominant traits have to occupy parts of the genome that are IBD-1 or IBD-2 among affected siblings and, if you assume complete penetrance, IBD-0 between affected siblings and unaffected siblings.
Penetrance is defined as “the proportion of individuals carrying a variant who also express an associated trait.”
How mapping using microsatellites works
For monogenic traits in large families it is possible to identify the locus based on extended linkage with surrounding polymorphisms. The methods for assigning grandparental haplotypes to grandchildren were originally worked out by the likes of Eric Lander and Mark Daly [Lander 1987, Donis-Keller 1987]. Though now done with SNPs, back in that day the mapping was done by genotyping microsatellites, a.k.a. short tandem repeats (STRs). A useful fact about these is that they are highly multi-allelic so you can uniquely identify grandparental haplotypes and identity-by-state (IBS) between siblings. To wit:
Fine linkage mapping requires either a very large family - like the huge Venezuelan family used to map the Huntington’s disease mutation [MacDonald 1993] or a multiple affected families who all have the same causal mutation, or at least have causal mutations at the same locus.
Linkage is quantified using a LOD score, which stands for “log of the odds”. A LOD score is calculated for every genotyped marker and is basically a log likelihood ratio. A marker’s LOD score is log10(likelihood of data if the phenotype is in linkage with this marker / likelihood of data if phenotype is not linked to this marker). Generally a LOD score of 3.0 is considered to be “some evidence” for linkage, and 4.0 is “stronger evidence”. This rule of thumb is based on multiple testing correction from the days when about 103 markers would be genotyped.
There are now >3,000 human diseases known to have monogenic inheritance [citation needed].

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.