Primer on PCSK9 genetics
The PCSK9 gene has emerged as a poster child of genetics and genomics. These days, almost any time you hear someone talk about the value of looking for loss-of-function mutations (as in this MIT Technology Review article), the value of human genetics for informing therapeutics (for instance in [Plenge 2013]), or the surprising power of “N of 1” studies (George Church’s talk at Harvard interview weekend), PCSK9 is sure to be mentioned. Here’s a primer on how this gene rose to fame in the past decade.
PCSK9 came to the attention of researchers a decade ago, when two missense mutations - S127R and F216L - were found to be associated with dominantly inherited familial hypercholesterolemia in two different families [Abifadel 2003]. Individuals with these mutations had high levels of low density lipoprotein (LDL or “bad cholesterol”) in their blood. The mechanism was originally unclear, but the authors speculated that the variants might cause a gain of function.
This speculation was soon vindicated: overexpression of PCSK9 - either the wild-type or mutant allele - in mice was shown to reduce LDL receptor levels and increase circulating LDL [Maxwell 2004], while Pcsk9 knockout in mice caused increased LDL receptor levels and reduced circulating LDL [Rashid 2005]. The knockout mice were also viable and fertile and seemed healthy.
Human genetics soon provided further validation: heterozygous nonsense mutations - causing a clear loss of function - were found in individuals with very low LDL [Cohen 2005]. Indeed, the association between less PCSK9 and less LDL isn’t just binary, it’s continuous - a whole spectrum of hypomorphic missense alleles of varying allele frequencies resulted in reduced secretion of PCSK9 and reduced LDL [Kotowski 2006]. The effects reported by [Kotowski 2006] weren’t limited to rare variants either, consistent with the fact that the PCSK9 locus has also turned up as a top hit in genome-wide association studies for LDL, demonstrating that common variants contribute too. (R46L may be the causal variant underlying much of the association in GWAS [Sanna 2011].)
All of this was exciting because, as cardiac geneticist Sekar Kathiresan says, “If there’s one thing in medicine that’s certain, it’s that LDL causes heart attack”. And indeed, a followup study in a much larger cohort soon revealed that not only were full or partial loss-of-functions in PCSK9 associated with reduced LDL, which is but a mere biomarker, they were also associated with a true clinical outcome: heart disease risk [Cohen 2006].
By 2006, statins had already been on the market for 20 years, and some had already started to become available as generics, yet millions of people still had high LDL [Kidd 2006, Steinberg & Witztum 2009, Crunkhorn 2012], and heart disease was (is) still the number one cause of death in the United States. Could inhibitors of PCSK9 be the next generation of LDL-lowering drugs?
The enthusiasm for PCSK9 inhibition as a therapeutic strategy got a huge boost when one compound heterozygote - someone entirely lacking a functional copy of PCKS9 - was identified [Zhao 2006]. She had one nonsense mutation and, in trans, an in-frame deletion, ΔR97, that entirely prevented secretion of the protein. This individual had extraordinarily low LDL and no other obvious phenotype:
Despite having no immunodetectable circulating PCSK9 and an LDL-C of only 14 mg/dL, II.2 is an apparently healthy, fertile, normotensive, college-educated woman with normal liver and renal function tests (including urinalysis) who works as an aerobics instructor.
This was the kind of “N of 1” insight that George Church loves to tell aspiring biologists about: just one healthy individual lacking a gene is enough to prove you can live without it. (An additional knockout individual, homozygous for a nonsense mutation, was also identified in [Kotowski 2006], although no phenotypic data were reported.)
So what is PCSK9? It turns out that it is a secreted serine protease [Horton 2007]. The N terminus has a signal peptide that directs it to the ER, followed by a prodomain. Once the prodomain is cleaved, PCSK9 becomes active and binds and degrades the LDL receptor, LDLR. LDLR is found on the surface of many cells, chiefly hepatocytes, and binds and internalizes circulating LDL. Thus, less PCKS9 means more LDLR, means less LDL.
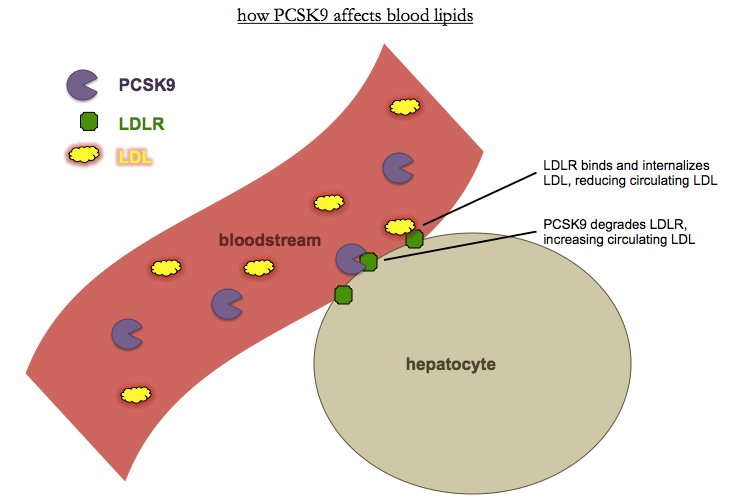
Together, all of this evidence made a brilliant case for therapeutics. It’s easier to develop inhibitors than activators, and here’s a gene whose loss is protective against the most common disease in the developed world, without any obviously deleterious side effects. Adding to that, there wasn’t just one loss-of-function allele but a full allelic spectrum, from homozygous or compound heterozygous loss of function, to heterozygous loss of function, to heterozygous partial loss of function, to wild-type, to pathogenic gain of function. It was, as David Altshuler and others would later observe, nature’s own dose-response curve [Plenge 2013].
Sadly, PCSK9 did not prove readily amenable to small molecule inhibition. Undaunted, Amgen developed a monoclonal antibody, evolocumab, which showed efficacy first in mice and non-human primates [Chan 2009] and later in humans. In parallel, Regeneron/Sanofi developed their own mAb, alirocumab, which showed outstanding results in Phase 1 and 2 trials [Stein 2012a, Stein 2012b]. The two companies were, at last count, locked in a bitter struggle to have their drug approved first - but as far as I’ve seen, all observers seem to agree that they will likely be approved, probably in 2015 (see e.g. Forbes, TheStreet). Other companies also have mAbs in development, and small peptides, siRNAs, and small molecules are in the works too, further back in the pipeline [Seidah & Prat 2012, see Table 5]. The continued desire for a small molecule is also presumably one motivator for continued work to identify other pathways which may lower PCSK9 indirectly and might be more readily druggable with small molecules - see for instance the story of SEC24A [Chen 2013].
If drugs against PCSK9 are indeed approved, it will be a proud day for human genetics. Loss-of-function mutations in several other genes have been reported to have beneficial phenotypes (some in a heterozygous, some in a homzygous state) - CCR5 and CXCR4 for HIV resistance [Samson 1996, citation needed], FUT2 for norovirus resistance [Thorven 2005], LPA, APOC3, and NPC1L1 for reduced LDL and/or heart disease risk [Ogorelkova 1999, Lim 2014, Pollin 2008, TG and HDL Working Group 2014, Jorgensen 2014, MIGCI 2014], ACTN3 for endurance athletic performance [MacArthur 2007], MSTN for strength and musculature [Schuelke 2004], SLC30A8 for reduced type 2 diabetes risk [Flannick 2014], and IFIH1 for reduced type 1 diabetes risk [Nejentsev 2009]. Some of these discoveries have already inspired (or in the case of NPC1L1, retrospectively validated) efforts to develop inhibitors. New human “knockouts” are being discovered all the time - see APOE earlier this year [Mak 2014]. Hopefully as we learn the loss-of-function phenotypes for more and more genes, we’ll have better and better ideas of what proteins to target with drugs.
Thanks to Brendan Bulik-Sullivan, Sisi Sarkizova, Sekar Kathiresan and Konrad Karczewski for catching mistakes and omissions.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.