Genetics in medicine 05: Cystic fibrosis
These are my notes from week 5 of Harvard’s Genetics 228: Genetics in Medicine from Bench to Bedside course, February 27, 2015. Lectures delivered by Dr. Leonard Sicilian and Dr. Sam Moskowitz.
Backgrounder
Cystic fibrosis is the classic example of a recessive disease. It is classic because it’s shockingly common: the ΔF508 mutation alone has an allele frequency of just over 1% in Europeans. This is a far higher allele frequency than natural selection would normally tolerate for any recessive disease mutation, especially one that, until modern medicine became good at managing symptoms, was lethal in early childhood [Quinton 1999]. It’s therefore speculated that this mutation simply must confer some sort of heterozygote advantage, and a lot of possible explanations have been proposed, including resistance to cholera toxin [Gabriel 1994], reduced risk of asthma [Schroeder 1995] and resistance to Salmonella typhi [Pier 1998], though no answer has emerged as conclusive.
CFTR encodes a chloride channel, and different genetic mutations cause a loss of its function by different mechanisms. Nonsense and frameshift mutations simply abolish the full-length protein. Splice site variants can reduce expression levels. ΔF508 causes the protein to be retained and degraded in the ER instead of transported to the cell surface, possibly by burying the di-acidic signal for cell surface localization [Wang 2004]. R334W affects the ability of chloride to pass through the channel. Finally, a whole class of missense variants including G551D affect the “gating” of the chloride channel.
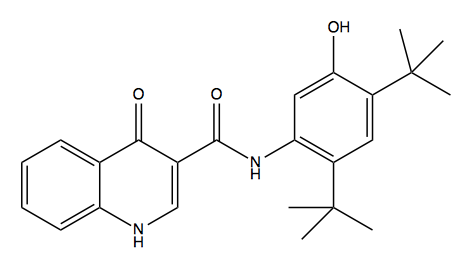
Above: ivacaftor, or N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide.
Vertex Pharmaceuticals, with the help $75M in funding contributed by the Cystic Fibrosis Foundation, developed the drug ivacaftor (Kalydeco™) which corrects CFTR gating defects. It received FDA approval in 2012 after clinical trials in compound heterozygous patients with G551D and a different mutation in trans [Accurso 2010, Ramsey 2011]. It was exceptionally effective in one G551D homozygote [Harrison 2013]. In 2014, ivacaftor’s indication was expanded to include eight other missense mutations associated with gating defects. Cystic Fibrosis Foundation has since sold its royalties on the drug for $3.3 billion, and in his 2015 State of the Union address, President Barack Obama praised ivacaftor as an example of what he hopes is a new wave of precision medicine drugs.
Without doubt, ivcaftor is a smashing success story. Yet even with its newly expanded indication, it is approved for only a few percent of CF patients. About 70% of CF patients have at least one ΔF508 allele [Castellani 2008]. These patients and Vertex Pharmaceuticals alike would be thrilled if ivacaftor were to prove effective for that mutation as well. The paper for this week [van Goor 2014] therefore sets out to quantify the degree to which ivacaftor corrects defects in cells expressiong CFTR with a panel of different genetic mutations.
Leonard Sicilian: CF in the clinic
CFTR protein regulates the composition and quality of liquid on the surface of the airway. Lack of functional CFTR dehydrates the cilia on the airway, impairing mucus clearance and creating an environment where bacteria can thrive. The CFTR gene was cloned in 1989 [Riordan 1989].
Children tend to be the face of cystic fibrosis, and indeed, a generation or two ago, there was no such thing as an adult CF patient. Projected median lifespan has risen dramatically, from 27 years in 1986 to 37 years in 2011, mostly due to better antibiotics and better nutrition. The number of adults with CF has risen dramatically, both because more patients survive childhood and because there is increased diagnosis of individuals who have less expressive mutations and a milder phenotype. Though we think of CF as a chronic debilitating disease, most adults with CF are employed and reasonably well-educated, and only about 15% of them are considered disabled. The usual measure of lung function is forced expiratory volume in 1 second (FEV1), which is the volume of air you can exhale in 1 second. By this metric, most patients are only mildly impaired in childhood (age ~7) but the proportion that are severely impaired rises to almost one third by age ~20. FEV1 is a predictor of mortality.
The progressive decline in respiratory function is attributed to something called “Cole’s vicious cycle”. Decreased mucociliary clearance in CF leads to bacterial infections, which causes inflammation, which leads to further insult of the airway, further impairing mucociliary clearance.
A variety of respiratory tract infections are common in individuals with CF. The most common in children with CF is S. aureus and the most common in adults with CF is Pseudomonas aeruginosa. MRSA is also a growing problem. To make matters worse, these infections are transmissible between CF patients, and outbreaks periodically occur. There was once a hospital that had an outbreak that killed 20% of their CF patients over about 18 months. Even though antibiotics are a mainstay in the treatment of CF patients, they are used with great caution, to avoid promoting antibiotic resistance.
Of these infections, P. aeruginosa is responsible for the greatest mortality burden. Chronic colonization by P. aeuroginosa is associated with far worse survival outcomes. It was proposed that aggressive treatment at the first infection with P. aeruginosa might prevent chronic infection, but this hasn’t proven to be the case.
Initial clinical presentation of CF is usually one or more of the following:
- Chronic sinopulmonary disease. Chronic (read: daily) cough and sputum production, repeated airway infections, airflow obstruction, abnormalities on chest X-ray, and sinus disease and/or nasal polyps. This can be seen with tomography as bronchiectasis, which is scarring of the airway / lungs.
- Gastriointestinal abnormalities leading to nutritional defecits. Pancreatic exocrine insufficiency, painful recurrent pancreatitis, intestinal obstruction (because the GI tract is unable to hydrate stool), and sometimes cirrhosis.
- Male infertility. The vas deferens can atrophy absent CFTR function, resulting in obstructive azoospermia. Sometimes adults are referred based on infertility and only later are determined to have CF.
CF patients have a dramtically increased risk of diabetes (due to pancreatic beta cell death), bone disease (due to poor nutrient absorption), and depression (probably secondary to receiving a grim prognosis). The prevalence of these conditions among CF patients rises with age, rising above 25% in one’s 30s.
CF diagnosis was traditionally made using sweat chloride levels, but increasingly, the diagnostic test is immunoreactivity for some other protein whose levels are upregulated in the absence of CFTR. Alternately, finding two genetic mutations in trans that are known to cause CF is considered sufficient for a diagnosis. Finally, diagnosis can also be made by measuring electrical potential in the nasal passage, which is altered in CF patients due to ion imbalance.
Not all CFTR mutations are created alike. We divide them into the following classes:
| class | severity | genetic basis | description |
|---|---|---|---|
| I | severe | Nonsense, frameshift | No synthesis of CFTR |
| II | severe | ΔF508 and some missense | Increased degradation of CFTR |
| III | severe | G551D and other missense | Gating defect. Protein is synthesized and localizes correctly but does not open and close. |
| IV | mild | Missense | Abnormal conductance |
| V | mild | Splice site | Reduced CFTR expression because not all transcripts are viable |
Wild-type CFTR is incredibly efficient, transporting about 10,000 Cl- ions per second. It is believed that the disease presents only when you drop below about 20% of wild-type function.
In addition to directly transporting chloride, CFTR has some other native functions as well. It also down-regulates transepithelial sodium channels, regulates calcium-activated chloride and potassium channels, and influences inflammation, cell maturation, ion transport and cell signaling.
Treatment of bronchiectasis includes i dentifying the bacteria that have colonized the airway, and aggressively treating acute exacerbations with intravenous antibiotics. Outside of acute exacerbations, doctors prescribe aerosolized antibiotics, delivered by an inhaler, for chronic suppression of infections, typically in a one month on, one month off region. The month off is intended to give non-resistant bacteria a chance to grow again and outcompete resistant bacteria, as resistance is presumed to come at some cost in terms of proliferation efficiency. However, in the long term, this regimen does lead to resistance traits becoming fixed in the bacterial population. Mucolytic drugs such as dornase alfa are also used to help clear mucus. Dornase alfa is actually a recombinant DNAse, and it reduces mucus viscosity by digesting DNA from dead cells. Hypertonic saline is used to extract moisture from lungs into mucus by osmotic pressure. This treatment was actually discovered after patients reported that their symptoms were partially alleviated after spending time in the ocean. Anti-inflammatories such as systemic or inhaled steroids can help airway flow, but have too severe of side effects for long-term use. NSAIDs have more limited benefits, and only at huge doses that are toxic to the kidney.
GI symptoms of CF can be treated by supplementing pancreatic enzymes, adding vitamins that are malabsorbed, and having a high-calorie diet to compensate for low nutrient absorption. If diabetes develops, it is treated with insulin, with no dietary restrictions (because of the need to get as many calories as possible). Laxatives are used to improve stool passage.
Inhalable adenovirus-vectored wild-tpe CFTR was tested as a gene therapy for CF. It showed transient benefits for a few weeks, which ended when the adenovirus was rejected by the host immune system. Interest in gene therapy for CF declined after a man named Jesse Gelsinger died of a severe immune reaction to an adenoviral vector used in a gene therapy clinical trial for a different disease [Sibbald 2001].
In the clinical trials for ivacaftor, the first sign of efficacy was a reduction in sweat chloride levels. Untreated patients had a progressive decline in FEV1, while treated patients’ FEV1 improved by about 15%. Treated patients also had improvements in several other endpoints, including quality of life [Ramsey 2011].
More recent clinical trials have tested ivacaftor, in combination with a new compound called lumacaftor, in ΔF508 homozygotes - see [Boyle 2014] and the CFF press release and the Vertex press release. FEV1 improved by 4.3 to 6.7%, an amount which is highly statistically significant in the clinical trial, but which may represent a small, perhaps even borderline imperceptible, benefit to any individual patient.
Sam Moskowitz: Q&A
The relationship between CF and weather is complicated. Cold, dry weather can cause bronchospasms, but hot, humid air tends to contain more particulates. In addition, CF patients are at risk for hyponatremia when it’s very hot out and they sweat a lot, because they lose a ton of NaCl in their sweat. Parents of children with CF are advised to leave salty snacks out and available at all times.
Genotype is highly predictive of pancreatic phenotype - about 95% of people with the “severe” alleles according to the above table have pancreatic insufficiency. Genotype is less predictive of pulmonary phenotype - for instance, there is a huge range of severity of respiratory symptoms among ΔF508 homozygotes.Age at first P. aeruginosa infection is more predictive of pulmonary decline. TGFβ receptor genotype is believed to have modest effects on lung phenotypes in CF [Drumm 2005].
Participating in CF clinical trials requires a rigorous schedule of clinical visits which can be hard for people who work.
Infection control messaging for CF patients has grown more strident over time, to the point that CF patients are even advised not to associate with other CF patients, due to the potential for transmission of antibiotic-resitant bacteria. This has been driven by a series of outbreaks, most famously an outbreak of Burkholderia dolosa at Boston Children’s a decade ago. The Cystic Fibrosis Foundation has a rule that no more than one CF patient can attend any CFF event. Hospitals have special protocols for cleaning rooms between when CF patients come in.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.