Organic chemistry 14: Unimolecular beta elimination - carbocation rearrangements
These are my notes from lecture 14 of Harvard’s Chemistry 20: Organic Chemistry course, delivered by Dr. Ryan Spoering on March 4, 2015.
Unimolecular beta elimination (E1)
Suppose you have tert-butyl bromide. In a solvent that contains a nucleophile, this will slowly undergo SN1, with dissociation to the carbocation being the rate-limiting step. If the solvent has no nucleophile, then when the Br dissociates, the Br- itself becomes the only nucleophile available. it will therefore attack and steal a hydrogen from one of the methyl groups:
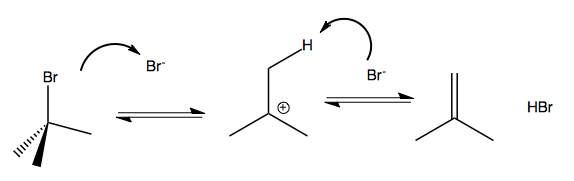
This reaction is reversible, and whether the reactant or products are more favorable depends on specifics. If you want it to run forward, you can heat it, because it turns out that the products are more entropic. If you think about ΔG = ΔH - TΔS, the higher T results in a negative ΔG, driving it forward.
Here is another example:
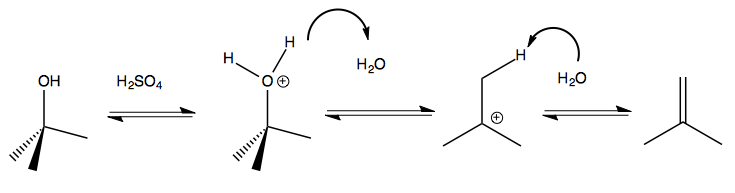
As a general rule, if you have something that can form a carbocation, and no other nucleophile is available, then E1 is the form that a reaction will take.
Regioselectivity
Saytseff’s rule says that more substituted alkenes are more favorable. Thus:
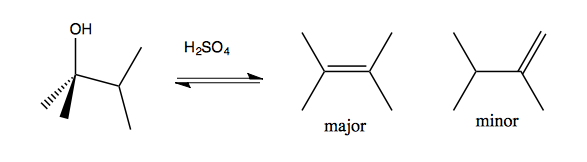
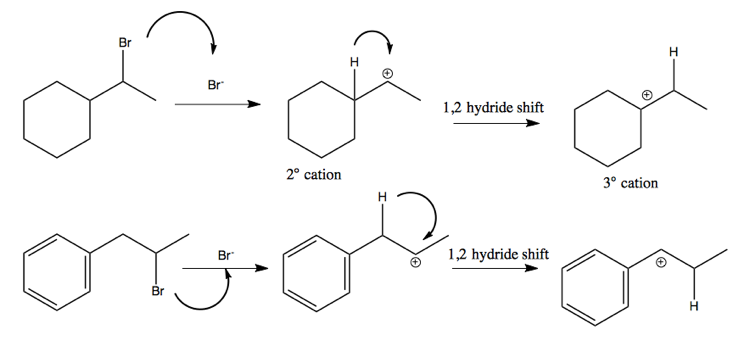
Carbocation rearrangements
1,2 hydride shift
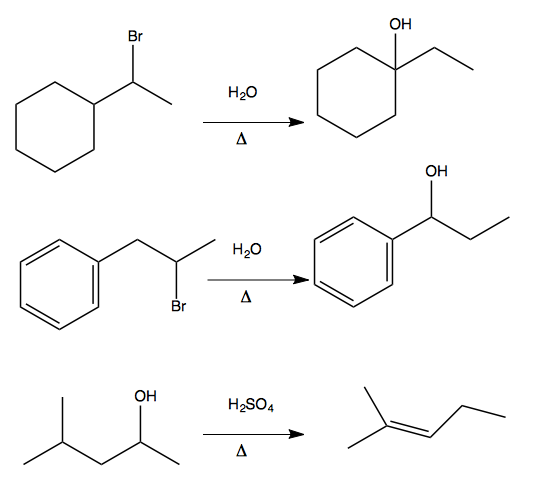
The above examples each proceed via a 1,2 hydride shift. Here are two examples of how this proceeds.
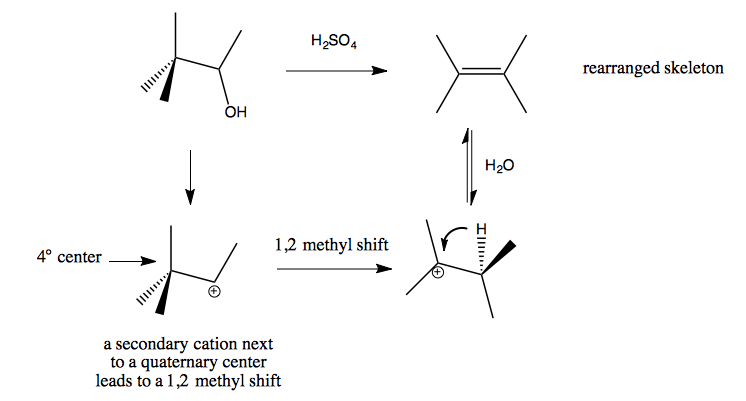
1,2 alkyl shift / Wagner-Meerwein Rearrangements
Here is one example:
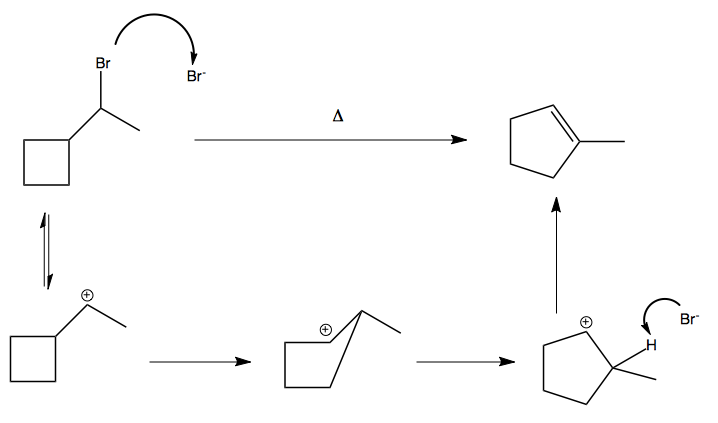
Another instance where this occurs is if you heat a strained ring:
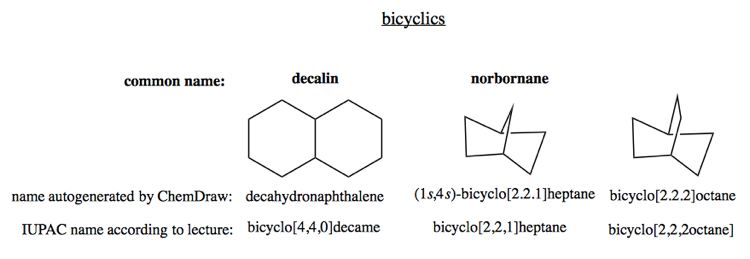
Bicyclic compounds
Bicyclic compounds can be something as simple as decalin, or they can be bridged bicyclics such as norborane.
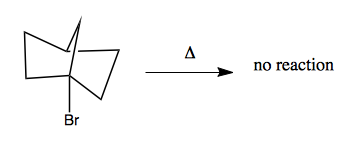
These moleules are very conformationally constrained. If you have a halide at a bridgehead, and you add heat, you get no reaction:
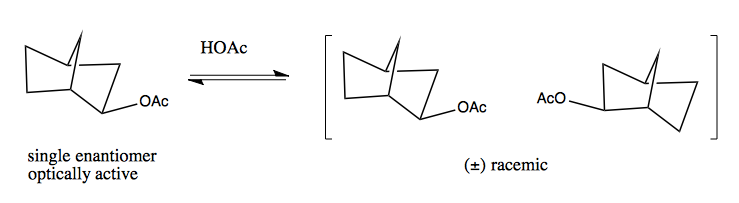
Here is a bicyclic where a single enantiomer will convert to a racemic mixture. You have to think hard about the mirror image to realize that the two forms shown at right are enantiomers. If you simply flip the -OAc group down instead of up, that actually forms a diastereomer, not an enantiomer (i.e. it is not a mirror image).
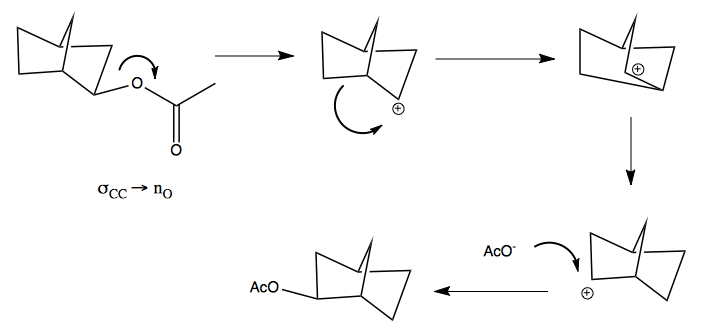
Here is the mechanism by which it interconverts. This was controversial for many years.
MPTP: a didactic tale
Carbocation rearrangement is also the mechanism behind the didactic tale of MPTP, which I’ve also told here. A college student in the 1970s wanted to synthesize meperidine (Demerol™), an opioid painkiller. He found an old paper describing the synthesis of a molecule called MPPP (“Bad Idea™”), which was described as having meperidine-like properties. He syntheized it and injected himself with it for several months, while also selling it to others.
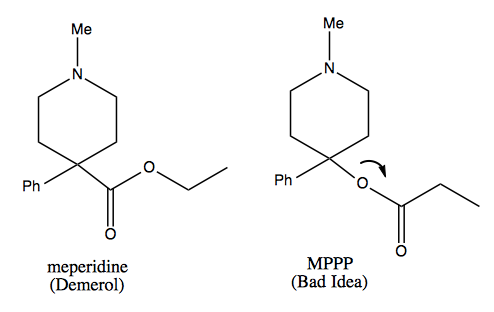
A synthetic chemist can recognize why MPPP is a bad idea, as follows.
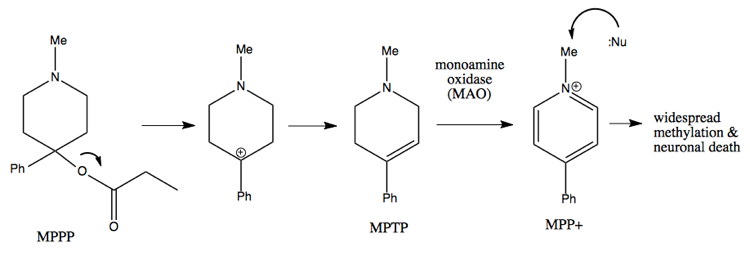
The quaternary center at the bottom of the ring in either molecule is a difficult thing to make, but in meperidine at least it is stable. In MPPP, the oxygen is a good leaving group, so it leaves a tertiary benzylic carbocation, which is the best kind of carbocation. This results in formation of a double bond. In dopaminergic neurons, that molecule is then converted by the enzyme monamine oxidase into the molecule at far right, which will add methyl groups to virtually any nucleophilic molecule in the cell. This results in massive covalent modification of things inside the cell, leading to neuronal death. The guy who was injecting himself with it presented in the emergency room with advanced parkinsonism, and later died.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.