Organic chemistry 28: Aromaticity - electrophilic aromatic substitution
These are my notes from lecture 28 of Harvard’s Chemistry 20: Organic Chemistry course, delivered by Dr. Ryan Spoering on April 17, 2015.
Electrophilic substitution of aromatics
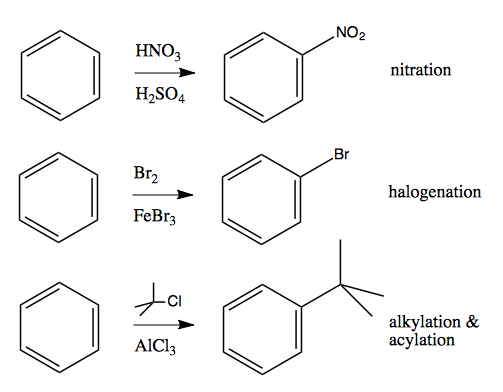
Today we’ll discuss several electrophilic ways of substituting a hydrogen on an aromatic ring, for instance, nitration, halogenation and alkylation / acylation:
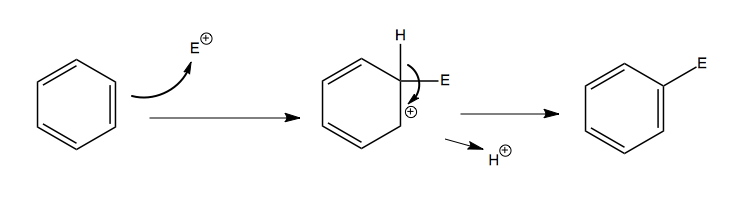
The general mechanism is as follows. E+ denotes an electrophile.
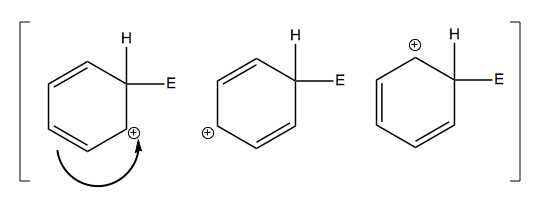
In the middle step, we’ve broken the aromaticity, which is tremendously energetically uphill. However, the species at center is stabilized by the fact that it has three resonance structures:
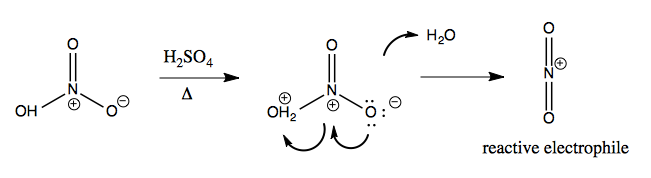
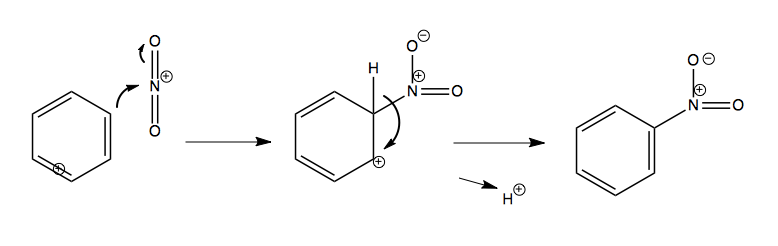
Nitration
Nitration of an aromatic is accomplished by first obtaining a reactive elctrophile NO2+:
And then combining it with an aromatic:
Halogenation
Halogenation is accomplished by first mixing the halogen with a Lewis acid such as AlCl3 or FeBr3 to make a reactive electrophile:
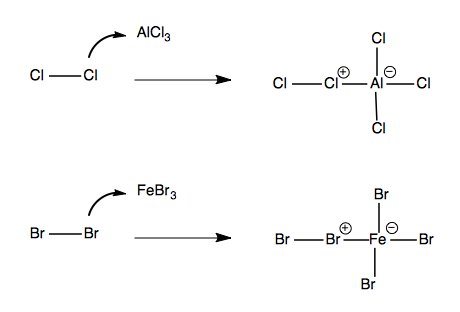
And then reacting this electrophile with an aromatic:
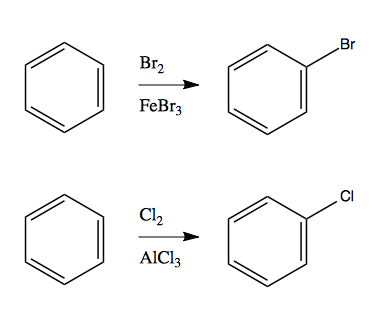
Friedel-Crafts alkylation
Friedel Crafts alkylation relies on a similar principle as the halogenation reaction above, but uses Lewis acid to steal the halogen from an alkyl halide, thus making a carbocation that can act as a reactive electrophile:
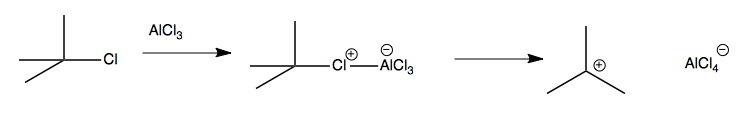
Once you have the carbocation, it can attack the aromatic ring:
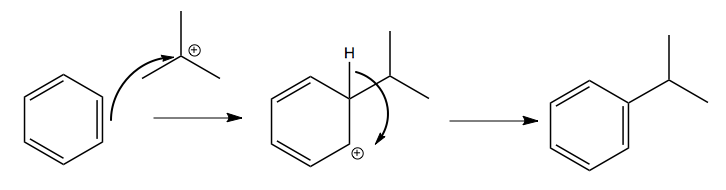
Friedel-Crafts acylation
Friedel-Crafts acylation is again similar, but uses the Lewis acid to create an acylium ion as the electrophile:
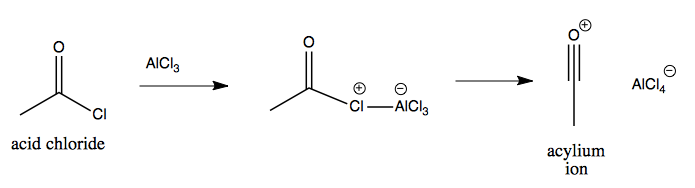
This acylium ion then reacts with the aromatic:
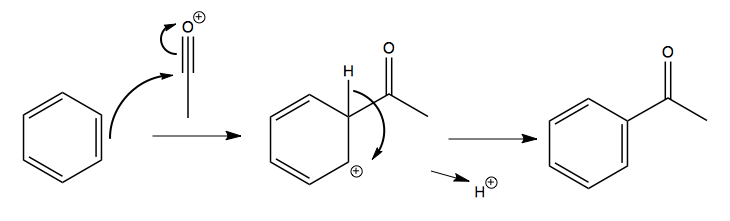
Multiply-substituted aromatics
Para, meta, and ortho positions
So far everything we have done is starting with benzene, thus making a singly substituted aromatic ring. What if we want to start with a substituted aromatic and add a second or third group?
In a substituted aromatic, the different carbon positions have names according to how far they are from the substituted group. Consider toluene:
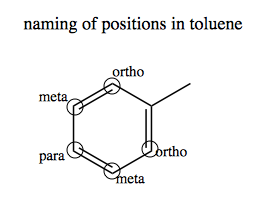
If we nitrate toluene, it will get nitrated only on the para carbon. If we further heat it, it will nitrate on the ortho carbons as well, but not on the meta carbons. Toluene triply nitrated on both ortho and the one para carbon is none other than the explosive TNT:
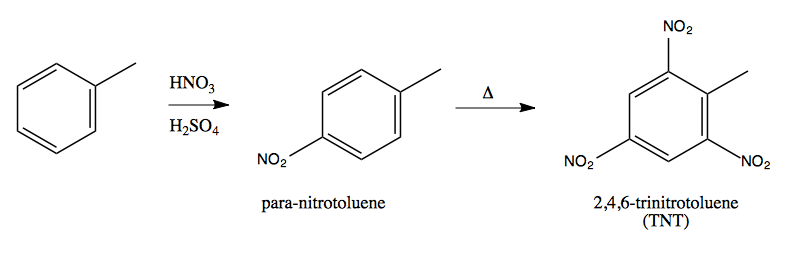
The preference for para, then ortho carbons over meta carbons can be understood by drawing the formally charged intermediates of these reactions and their resonance structures:
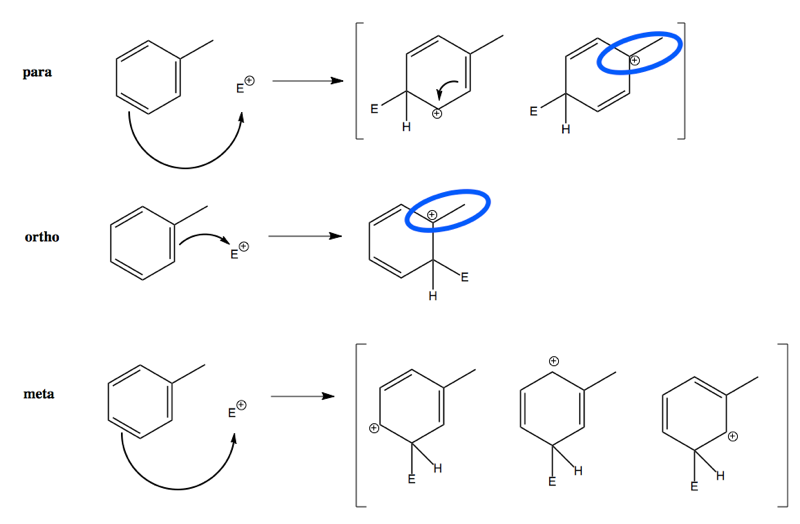
In the para substitution, there is a resonance structure wherein one of the two structures has stabilization by having the carbocation be adjacent to the methyl group (highlighted in blue). In the ortho substitution, there is just one structure, and it also has this stabilization (again in blue). In the meta substitution, there are three structures but none of them is able to benefit from sharing positive charge with the methyl group.
This lesson is a useful reminder of why it’s useless to draw benzene with a circle in the middle. Only by drawing individual double bonds in benzene can one understand the preference between para, ortho, and meta carbons.
Effects of substitution
Substitutions on an aromatic ring can decrease or increase the propensity to acquire another group. This depends upon the spectrum from electron-donating to electron-withdrawing groups:
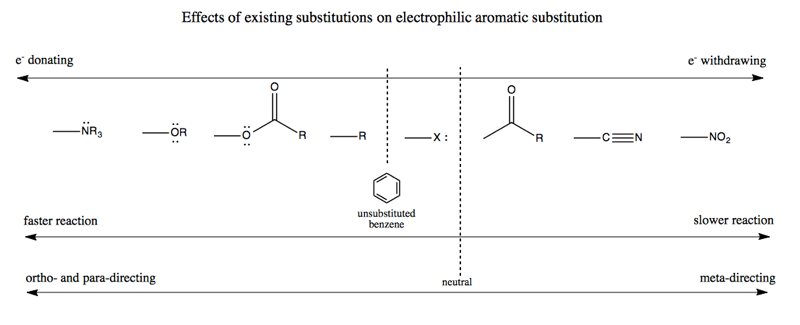
Note that “ortho- and para-directing” is often abbreviated “o,p-directing”.
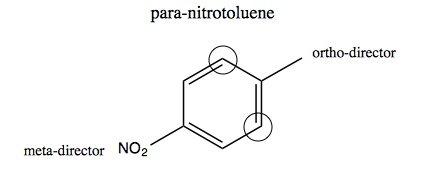
With this framework in our quiver, we can now go back and understand why heating para-nitrotoluene results in 2,4,6-TNT as opposed to some other configuration. In para-nitrotolune we’ve already added the first NO2 group in the lower left, which is the para position with respect to the methyl group. NO2 is a meta-directing group (see above). We also still have the methyl, which is a para- and ortho-directing group (it counts as -R in the chart above). The two circled carbons in the graphic below are meta with respect to the NO2 meta director, but ortho with respect to the ortho-directing methyl. Therefore the two groups are in perfect agreement that any additional groups should be added at these two positions rather than at the other two, non-circled carbons:
When making multiply substituted aromatics, it is critical to think in advance about the ortho-, para- versus meta-directing properties of the groups you want to add. First, here are two useful reactions you should know - nitro group reduction and Clemmensen reduction:
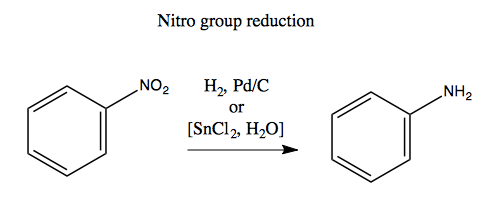
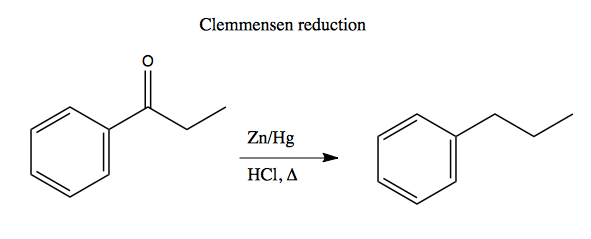
Now, suppose you had this retrosynthesis problem:
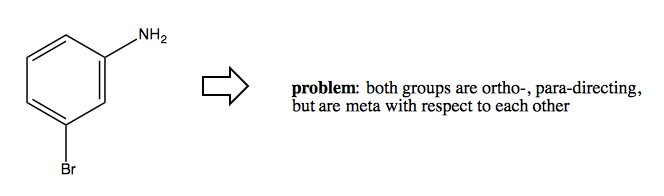
You want to add Br and NH2 groups to a benzene, but you have the problem that both of these groups are ortho-, para-directing, yet they are each in the meta position relative to one another. Thus, as soon as you add one, it is no longer possible to add the other.
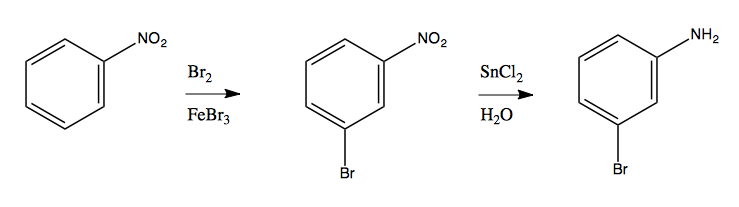
To solve this problem, you have to first add a NO2 group, which is meta-directing. Then you can add the Br group in the meta position using Br2 and FeBr3. Now that you have substitutions in the two positions you want, you can use nitro group reduction (with SnCl2, pictured above) to reduce the NO2 to NH2. Like so:

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.