The powerful protective effect of G127V
John Collinge’s plenary talk at Prion2015 in Colorado last month got off to a gradual start. He spent the first half hour reviewing the history of the kuru epidemic in Papua New Guinea, showing photos of rainforested hillscapes and videos of interviews with Fore people recalling the prion disease that, at its peak, wiped out 2% of their population annually. Then he segued into genetics, talking about his group’s finding, now six years old, that the G127V missense variant in PRNP underwent massive positive selection during the kuru epidemic [Mead 2009]. From its origins ~10 generations ago (the estimated time to most recent common ancestor of Fore G127V chromosomes) the variant rose to a 7% allele frequency in the most heavily affected regions, with not a single person carrying the variant falling ill with kuru.
Above: the thatched-wall BL2 prion room constructed in the Fore region, complete with a hand-powered centrifuge, from Dr. Collinge’s talk at Prion2015.
Kuru is an old story that, in Sonia’s words, “we didn’t expect to produce another chapter”. But to our surprise, halfway through the talk, Dr. Collinge launched into some brand new findings, which are published today in Nature [Asante 2015]. (See also Glenn Telling’s commentary and Boer Deng’s Nature News story). MRC Prion Unit generated two lines of transgenic mice expressing 127V HuPrP at ~1x and ~2x wild-type levels and tested their susceptibility to a variety of human prion isolates. Amazingly, when 127V was the only PrP being expressed, the mice evinced absolute resistance to all tested prion strains including four kuru isolates and several sporadic, iatrogenic, and variant CJD isolates. When the transgene array was heterozygous, in trans to a wild-type 129M or 129V HuPrP allele, the resistance was still nearly complete, with only vCJD prions achieving a ~50% attack rate.
Skeptics may note that the 127V transgenic mice in this study only expressed 1-2x wild-type levels of PrP, which is low compared to some transgenic lines. For instance, the first human PrP transgenic mice ever created, Tg152 [Telling 1994], which make a cameo in this study, express 6x wild-type levels of PrP. In general, higher PrP expression levels make for a more rapid disease course [Fischer 1996], and HuPrP knock-in mice, which express only 1x the wild-type level of PrP, exhibit incomplete attack rates for multiple human prion strains [Bishop 2006, Bishop 2010], even though they lack the 127V variant. Some human prion strains - particularly some of the kuru isolates tested here - have incubation times that last the better part of the natural lifespan of a mouse even in humanized mice without the 127V variant (see Table 2a, in Asante 2015). Therefore, one might wonder this study is fully powered to discriminate between complete resistance to prion disease versus just very long incubation times. In this study, however, the most salient comparison is between homozygous Tg35 mice (2x levels of wild-type PrP) and homozygous Tg183 mice (2x levels of 127V PrP). For all prion strains tested, almost 100% of the mice expressing wild-type HuPrP became sick (and the few that did not still showed subclinical infections), while not one of the homozygous 127V mice became sick, nor showed any histological signs of subclinical infection. While no transmission studies were undertaken to assess the possibility of low levels of prion propagation (as was once done, for instance, to show that PrP knockout mice are completely resistant to prions [Bueler 1993]), the data are certainly consistent with total resistance to prions, and at a minimum, the data are unambiguous that the protective effect of the G127V allele is very, very strong — apparently far greater than that of the well-known protective effects of M129V or E219K heterozygosity [Palmer 1991, Shibuya 1998, Bishop 2009].
Before you get too excited, the bad news is you almost certainly don’t have the 127V variant in either copy of your PRNP gene. It is ultra-rare, absent from 60,706 people in ExAC, and as Dr. Collinge noted, has so far never been seen in any population besides the Fore. And in case you’ve been taken in by the past year’s media blitz about genome engineering, it’s also worth noting that our ability to deliver genome editing vectors to the human brain is emphatically not at the point where this information is useful for gene therapy approaches either.
Instead, the news about G127V will probably be most interesting if it can, in the long run, help guide us in designing a small molecule drug to mimic its protective effect. Such insights are not unprecedented. Consider the story of tafamidis. This drug, now licensed in the European Union for treating familial amyloid polyneuropathy (FAP), works by stabilizing transthyretin tetramers, preventing their dissociation into monomers, a requisite step on the pathway to formation of amyloid plaques. Years ago, when Jeff Kelly’s group at Scripps was first experimenting with small molecules to stabilize TTR tetramers, it got a major piece of human genetic validation for its therapeutic strategy from the discovery that a protective genetic variant, T119M, also stabilizes the tetramers [Hammarstrom 2001, Hammarstrom 2003]. There is an increasing recognition among some in the pharmaceutical industry that these sorts of “experiments of nature” from human genetics can provide the best possible guide to therapeutic development [Plenge 2013].
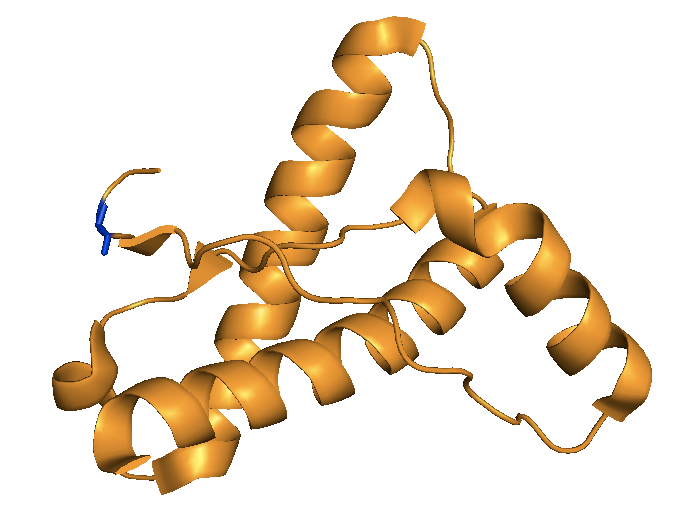
Above: the crystal structure of human PrP’s C terminus from [Antonyuk 2009] with residue 127G highlighted in blue. The protective variant changes this small, flexible glycine residue to a bulky valine residue.
But making sense of the mechanism of G127V’s protective effect will be anything but easy. After all, we’ve known for decades that 129MV heterozygosity is protective against sporadic CJD [Palmer 1991], and more generally that the co-expression of two incompatible PrP amino acid sequences inhibits prion propagation [Telling 1995], but leveraging that information for therapeutic design has not proven fruitful so far. PrP doesn’t give up its secrets as easily as TTR does. The C terminus crystallizes as a domain-swapped dimer [Knaus 2001] or sometimes with an intermolecular beta sheet [Antonyuk 2009] sometimes as a monomer [Lee 2010], and the N terminus appears totally unstructured. We still don’t really know how PrPC behaves on the cell surface, it doesn’t appear that pathogenic mutations necessarily destabilize the native state of the protein [Swietnicki 1998], and although the new paper [Asante 2015] claims that 129MV heterozygosity inhibits “homotypic protein–protein interactions”, the truth is that we still don’t really know, at any meaningful molecular resolution, how any the protective variants of PrP exert their effects. Years ago, the Prusiner lab embarked on an effort to use an in silico screen to conjure up molecules that would mimic the effect of dominant negative PrP genotypes [Perrier & Wallace 2000], and although they found two hits that were active in cell cultures, neither were as potent as other compounds that have subsequently been identified through phenotypic screens (EC50s of 15-35 μM compared to more like 1μM for IND24, cpd-b, and anle138b [Berry 2013, Kawasaki 2007, Wagner 2013]).
At Prion2015, Dr. Collinge briefly touched on structural studies that his group has undertaken to try to characterize the effect of the 127V variant. But it’s possible that the protective effect is not mediated by any change to the structure of PrPC. If 127V instead acts, say, as an antagonist of the PrP-recruiting surface of PrPSc, then it will be hard to figure out exactly how it works, since we still don’t know the structure of PrPSc.
To me, it is also still unclear whether the mechanism of G127V’s protective effect is fundamentally different from that of M129V or E219K. The new paper [Asante 2015] makes the following claim about G127V:
…the molecular basis of this effect is clearly distinct from that proposed for the well established HuPrP M129V polymorphism, which is protective against developing sporadic CJD only in the heterozygous state…
While it’s true that M129V’s greatest protective effect by far against sporadic CJD is in a heterozygous state, the evidence for other human prion strains is more mixed. For instance, Jean Manson’s experiments transmitting variant CJD to her HuPrP knock-in mice have suggested that 129VV mice may be even less susceptible to vCJD than 129MV mice [Bishop 2006]. In those experiments, both genotypes exhibited very limited susceptibility (only one mouse ever became clinically ill), so it’s hard to be certain of the conclusions, but this is at least some evidence that for variant CJD, the effect of M129V may in fact be allelic, or dose-dependent, in nature, just as G127V appears to be. So the mechanism by which G127V confers protection may not be entirely unprecedented.
While all of the above caveats are reasons to interpret the new findings with caution, the discovery that G127V has such a profound protective effect is still surprising and interesting. As the paper points out, the variant appears to be about as protective as wholesale deletion of PrP, which hasn’t proven to be the case for any other missense variant so far. The real test of the usefulness of this discovery will lie in whether it manages to produce yet another chapter — one in which it provides concrete insights to guide drug discovery.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.