Chemical biology 10: Chromatin and small-molecule modulators
These are my notes on lecture 10 of Harvard’s Chemistry 101: Chemical Biology Towards Precision Medicine course, taught by Dr. Stuart Schreiber on October 8, 2015. I missed this lecture while away at a conference, so these notes are based on the slides and reading materials.
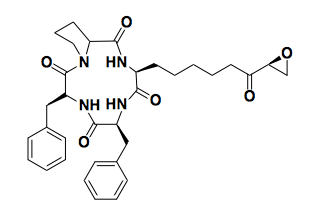
Above: trapoxin
Histone modifications were first noticed by Vincent Allfrey in 1964, who speculated they might be important, but it was years before we gained any understanding of how they regulate gene expression. The mammalian proteins that are responsible for reading and writing histone modifications did not start to be known until the 1990s.Trapoxin (above) is a fungal natural product that has anticancer properties — it can change the morphology of cells and make transformed cell lines behave more line normal cells. Trapoxin was known to prevent the de-acetylation of histones [Kijima 1993] but the putative histone deacetylase protein had not been identified. It was believed to be a covalent, irreversible inhibitor, and hypothesized to be an active site substrate mimetic. By conjugating it to a solid support, it was possible to pull down the protein and perform amino acid sequencing to characterize it [Taunton 1996]. This was the first histone deacetylase (HDAC), and it proved to be homologous to an already-characterized yeast protein.
How exactly did histones regulate gene expression? The histone code hypothesis held that:
Multiple histone modifications, acting in a combinatorial or sequential fashion on one or multiple histone tails, specify unique downstream functions.
The competing signaling network model of chromatin held that there was no unique specification of function, but rather that histone modifications worked simply through effective molarity. Two proteins that bound to histones with two particular modifications would come into proximity with one another when both modifications were present, proximity thus allowing those two proteins to interact. This is analogous to how cell signaling works, for instance, how phosphorylation of receptor tyrosine kinases creates a docking site for multiple proteins to interact and form a complex.
The development ChIP-seq made it possible to study histone marks genome-wide, helping to elucidate the ways that chromatin state specifies cell state. H3K27 methylation is found in inactive genes, while H3K4 methylation is found in active genes; famously, it was discovered that undifferentiated embryonic stem cells have many genes with both H3K27 and H3K4 methylated, a “bivalent” state which makes the genes “poised” to turn on as the cell differentiates [Bernstein 2006].
A challenge of precision medicine is to use small molecules that modulate chromatin to achieve transdifferentiation of cells in vivo. There have been some proofs of principle, for instance, in reversing HIV-1 latency so that dormant HIV becomes sensitive to antiretroviral drugs [Archin 2012]. And it’s been proposed that one could turn on fetal hemoglobin expression to treat sickle cell disease through HDAC modulation [Bradner 2010], and that HDAC3 inhibition can protect pancreatic beta cells from cytokine-induced apoptosis in a model of type 1 diabetes [Chou 2012]. What everyone wants for a drug is a molecule that turns non-insulin-secreting pancreatic cells into insulin-secreting beta cells.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.