Chemical biology 20: cancer stem cells
These are my notes from lecture 20 of Harvard’s Chemistry 101: Chemical Biology Towards Precision Medicine course, taught by Dr. Stuart Schreiber on November 19, 2015.
Last time we talked mostly about oncogene addiction and the molecular view of cancer. Today’s lecture is on the cancer stem cell view of cancer. Some people call these cells “tumor progenitor cells” or “persister cells”. Some readings for today are [Diamandis 2007, Dannenberg & Berns 2010, Hartwell 2013, Pattabiraman & Weinberg 2014].
The four main mechanisms of cancer resistance to drugs, and strategies for overcoming them are:
| mechanism | strategy |
|---|---|
| mutations in a drug target that prevent the drug from binding | new drug (e.g. ponatinib) that binds the mutated target |
| new pathway is upregulated (e.g. acquisition of mesenchymal state) | combination therapy with a drug that targets the upregulated pathway |
| upregulation of a drug efflux mechanism | combination therapy with a drug that blocks efflux |
| cancer stem cells | new drug targeting cancer stem cells |
Let’s talk more about what we mean by “cancer stem cells” being a resistance mechanism. The theory is that in many tumors, only a subset of cells have unlimited replication potential, and they are ultimately responsible for maintaining tumor mass. These cells have stem-like properties, and are resistant to many cancer drugs.
We define stem cells on the basis of two unique properties:
- Apparently infinite renewal capacity. This is in contrast to most cells, which can divide perhaps tens of times and then hit the Hayflick limit and become senescent.
- Ability to divide asymmetrically. Stem cells can divide such that one daughter cell is still a stem cell, while the other daughter cell begins to differentiate.
We’ll discuss the example of hematologic cancers. Hematopoeitic stem cells (the origin of blood) were discovered by irradiating mice to eliminate their hematopoietic system, and then seeing which cells were able to reconstitute their hematopoeitic system. Only HSCs could do this. Leukemic stem cells were discovered by doing the analogous experiment using leukemia cells: shockingly to people at the time, who thought leukemia was purely clonal, it turned out that about 1 in 100,000 leukemia cells had the ability to reconstitute a mouse’s hematopoeitic system. When these cells were characterized, they had all the markers of stemness. Once this was accepted, people started asking if the same was true of solid tumors. They tested this by labeling individual tumor cells with fluorescent markers, and were able to track how a single cancer stem cell could give rise to a huge swath of a tumor. A couple of relevant perspectives on all this are [Abbott 2006, Baker 2012] and it appears that three of the original articles on lineage labeling of solid tumor stem cells are [Driessens 2012, Chen 2012, Schepers 2012].
So how do we translate this insight into therapy? You could hypothesize that if you only kill the cancer stem cells, that would be sufficient to cut off the ultimate growth potential of cancers, but the bulk of the cancer would not immediately decrease. That is a challenging hypothesis to test in a clinical trial, because without debulking the tumor it looks like there was no clinical benefit. So in practice, any drugs targeting cancer stem cells will need to be administered in combination with conventional debulking therapies.
When cancer stem cells give rise to cancer relapse, the relapsed cancer is usually much more aggressive. Why? In one landmark experiment, human glioblastomas were transplanted into mice, and then later resected, with or without radiation. When it was resected without radiation, the relapse that eventually emerged had a low percentage of cancer stem cells. When it was resected with radiation, the relapse had a high percentage of cancer stem cells. It is obvious that radiation therapy here is increasing the proportion of cancer stem cells in the tumor. It may also be the case that it is actually increasing the absolute number of cancer stem cells, and therefore actively doing harm. That’s because when the tumor is intact, the cancer stem cells are limited in growth by the need to compete with the tumor bulk for nutrients, but when the tumor bulk is killed off, the cancer stem cells are more able to proliferate. Not sure but I think this part of the lecture might be referring to [Bao 2006].
Leukemia stem cells (LSCs) cannot be studied in non-adherent culture. LSCs, like HSCs, have an obligate need for physical contact with bone marrow stroma. They can be grown in the lab on primary bone marrow tissue. This points to a vulnerability that might be targeted therapeutically. For instance it was recently reported that the PPARγ agonists rosiglitazone and pioglitazone, which are approved for the treatment of diabetes, induce stroma to acquire a more adipose state that is less hospitable to leukemia stem cells [Prost 2015]. There is a whole body of literature on the fact that lovastatin also has anti-leukemia properties, though this does not work in vivo because lovastatin (and all other statins currently available) are highly liver-tropic, so they don’t reach high enough concentrations in bone marrow to be effective.
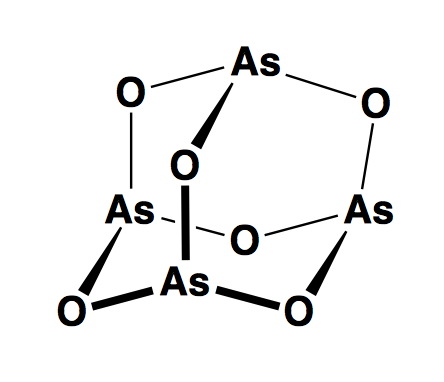
Of course, it is also a strategy to directly target the cancer stem cells. For instance there is new evidence from small molecule screening that neurotransmission pathways are surprisingly important in neural stem cells and that some of these pathways might be able to be targeted therapeutically in glioblastoma [Diamandis 2007]. Also there is the PML paper we discussed in section last week [Ito 2008]. Arsenic trioxide (As2O3) actually has the shape of adamantane:
One last concept that is fairly new is immune therapy. Cancer cells express mutated proteins on the cell surface, and the immune system is able to recognize these neoantigens and attack the cancer cells. But tumor cells often massively overexpress PD-L1 (programmed death 1 receptor ligand), which binds to the programmed death 1 receptor (PD-1) on the T cells, shutting down the immune response. So a new therapy is to use monoclonal antibodies against either PD-1 or PD-L1 to block this interaction. There are now approved antibodies for both of these targets. One recently approved drug, the monoclonal antibody pembrolizumab, targeting PD-1, was in the news recently for its role in Jimmy Carter’s so far very successful treatment regimen for metastatic melanoma of the brain.
So far, these sorts of immune therapies seem to be very effective at preventing relapse, but they are relatively new and we don’t yet have many years of followup. One very hopeful note about these therapies is that, opposite to all other cancer drugs, they are actually more effective in more advanced, late-stage cancers. That’s because the more heavily mutated cancers, which are better at evading all other therapies, have accumulated more neoantigens, and so are more readily recognizable to the immune system.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.