Huntingtin becomes the huntingted
According to an announcement earlier this morning, an gene silencing therapy against the huntingtin (HTT) gene has succeeded in lowering mutant huntingtin levels in patients’ cerebrospinal fluid, while also showing a good safety profile. It’s not an approved drug yet, nor is there yet direct evidence of clinical benefit — clinical trials will continue and it will probably be a few years before we know for sure if the drug is helping patients. But today marks a big step forward for the prospects for therapy of Huntington’s disease. And with this news come reasons for optimism about treatment of neurological disease generally, including prion disease.
This morning’s announcement comes from Ionis Pharmaceuticals and Roche, two companies that partnered in 2013 to bring an ASO against huntingtin to clinical trials. Ionis announced in October 2015 that they had dosed the first patient. That announcement, and today’s encouraging results, concern a Phase 1/2a clinical trial in Canada, Germany, and the U.K., led by Dr. Sarah Tabrizi. The trial enrolled a small number of patients, and started with a low dose that then escalated. The main goal of the trial was simply to assess whether the drug was safe enough to move on to a larger trial at a therapeutic dose in order to establish efficacy, but it also provided an opportunity to measure biomarkers to figure out whether the drug was having its intended effect — more on that in a moment.
The reason today’s news is so exciting is that the gene silencing therapy in question is an antisense oligonucleotide (ASO). Let’s pick that apart. Oligonucleotide means that the drug is a short sequence of nucleotides, the building blocks of DNA and RNA. Antisense means that the drug is the reverse complement of a nucleotide sequence found in the huntingtin gene itself. G pairs with C and A pairs with T, so to translate a sequence into its reverse complement, make it backwards and then replace G with C and vice verse, and A with T and vice versa. For instance, TAG is antisense to CTA. Because the sequence of the drug is reverse complement to the huntingtin RNA, the two will bind together, just like the two helices of DNA bind together. This binding, creating an unnatural double-stranded RNA molecule, causes an enzyme called RNAse H to come cut the RNA in half [Wu 2004]. Thus, when you treat with the drug, the amount of huntingtin RNA goes down, and therefore the amount of huntingtin protein, translated from that RNA, goes down as well.
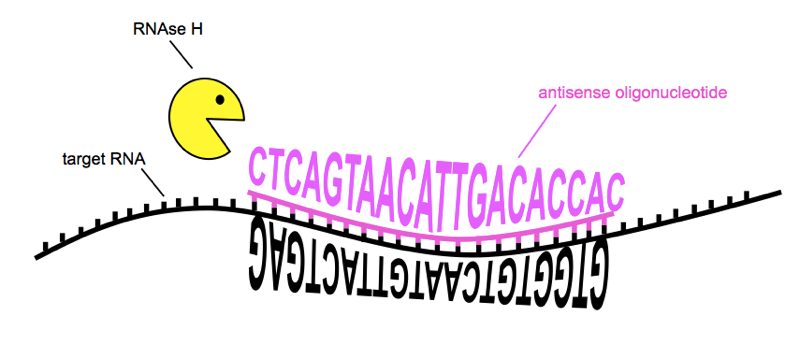
How ASOs reduce the amount of a target RNA. Sequences of RNA and ASO are from [Kordasiewicz 2012]
The great promise of ASOs lies in the fact that you can program the sequence of nucleotides to target almost any gene you want. So if you can design an ASO to successfully lower huntingtin, then it’s relatively straightforward to design an ASO to lower any other gene of interest. That doesn’t mean that drug development is easy — there are still lots of other obstacles to getting a new molecule into clinical trials and showing that it works — but it should certainly help. ASOs as a technology have been around for over 25 years, and for much of that time there’s been a lot of hype about how many different diseases out there could potentially be treated, by designing an ASO against the gene that causes each one.
A few relatively recent advances have made this dream finally start to seem near at hand. First, Ionis has developed new chemical modifications to the nucleotide backbone that make the molecules much more stable and potent than the “1st generation” ASOs of decades past [Bennett 2017]. Second, the approach to dosing for brain diseases has improved. It was previously thought that you needed to gradually infuse drug into the cerebrospinal fluid, so mouse studies were done with tiny pumps implanted in the brain [Nazor Friberg 2012] and there were human trials where a pumping in a single dose took place over 11 hours [Miller 2013]. Then, a few years ago, Ionis did a study with one of its ASOs that found that the drug was actually more potent (and still safe) if the whole dose was delivered in one shot [Rigo 2014]. Third, new evidence from humans and non-human primates over the past few years shows that ASOs dosed intrathecally — injected into the base of the spine — distribute and achieve activity throughout a wide swath of brain regions [Kordasiewicz 2012, Rigo 2014, Finkel 2017, DeVos 2017]. So it looks like the drugs actually go where they need to go to treat a lot of diseases that people care about.
The excitement about ASOs crystallized about a year ago, when FDA approved nusinersen, an ASO drug for spinal muscular atrophy (SMA), a previously untreatable genetic disease. Nusinersen proved remarkably effective even after symptom onset [Finkel 2016, Finkel 2017], and appears to have had even more dramatic effects when given preventively. It has become the first disease-modifying therapy for any neurodegenerative disease, and that’s a really big deal.
Still, the analogy is imperfect between that drug for SMA and the drug you’d want for many other neurodegenerative diseases. First, SMA is predominantly a disease of the spinal cord, and the drug was dosed into the intrathecal space (at the base of the spine), where it would have a relatively easy time reaching the spinal cord. So the fact that nusinersen proved effective for SMA didn’t necessarily prove that ASOs could be effective for diseases that primarily affect the brain, which is further from the intrathecal space. Second, what nusinersen actually does by binding its target RNA is to cause an increase in the amount of a good protein that SMA patients need, whereas in many neurodegenerative diseases, what you want is a decrease in the amount of a bad protein that causes the disease. So although there was lots of evidence in animal models that ASOs are good at lowering a protein of interest in the brain, and there was an approved drug for humans (mipomersen) that lowers a protein of interest in the liver [Raal 2010], there wasn’t quite any direct evidence that an ASO could lower a protein of interest in the human brain.
That’s why the new results for huntingtin are so exciting. The ASO tested in this clinical trial really needed to reach the brain (not so much the spinal cord) and what it needed to do in the brain was reduce huntingtin protein. It’s a very close analogy to what a drug in prion disease would need to do. And although we may not know for a few years yet whether the ASO was clinically effective (whether it improved the way patients feel or function), the information in this press release suggests that the drug did get to the brain, and it did reduce huntingtin. So: lowering a disease-causing protein in the brain appears to be possible.
The huntingtin ASO, like nusinersen, was dosed intrathecally, through a lumbar puncture. As explained here, the dosing was monthly, and the trial was randomized (some patients got placebo). The fact that the drug is delivered through a lumbar puncture makes it easy to also take patient cerebrospinal fluid to measure a biomarker. A couple years ago, partly in order to be able to measure the effect of a drug like this in clinical trials, Dr. Ed Wild and his team developed a method for measuring mutant huntingtin (mHTT for short) in cerebrospinal fluid [Wild & Boggio 2015]. Based on the press release, this was presumably the biomarker measurement they did in this clinical trial to determine that the ASO had successfully done its job at the molecular level. The press release has a quote from C. Frank Bennett, a scientist at Ionis, saying that “The dose-dependent reductions of mHTT we observed in the study substantially exceeded our expectations”.
We’ll look forward to hearing the full details about this trial hopefully sometime next year. But today’s news is one step forward toward the idea that for brain diseases caused by a misbehaving protein, it is possible to design a drug to lower the amount of that protein in the brain. To me, that’s a great cause for hope.
Update 2018-05-22
The underlying data were finally announced in a press release and at a conference in March. At the two highest doses, the ASO lowered mutant huntingtin by 40% in CSF. And the drug was safe: among the 46 people enrolled in the trial, there were no serious adverse events.
This is incredibly exciting news, but it’s also important to remember that this doesn’t yet prove that the drug is clinically effectve. The results from animal models suggest that 40% reduction in mutant huntingtin should be beneficial [Kordasiewicz 2012], but it is still conceivable that in humans, 40% might prove to be too little knockdown, or even that it might prove to be too much knockdown (because it’s essential to have some huntingtin around). To figure out whether the drug is effective at slowing progression or improving symptoms of HD, the expectation is that next there will be a pivotal trial in a larger number of HD patients. We’re still awaiting details of what that trial will consist of.
For more perspective on what the results mean, see HDBuzz’s post on the conference announcement and Q&A about the press release and next steps.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.