The variants you see and the variants you don't see
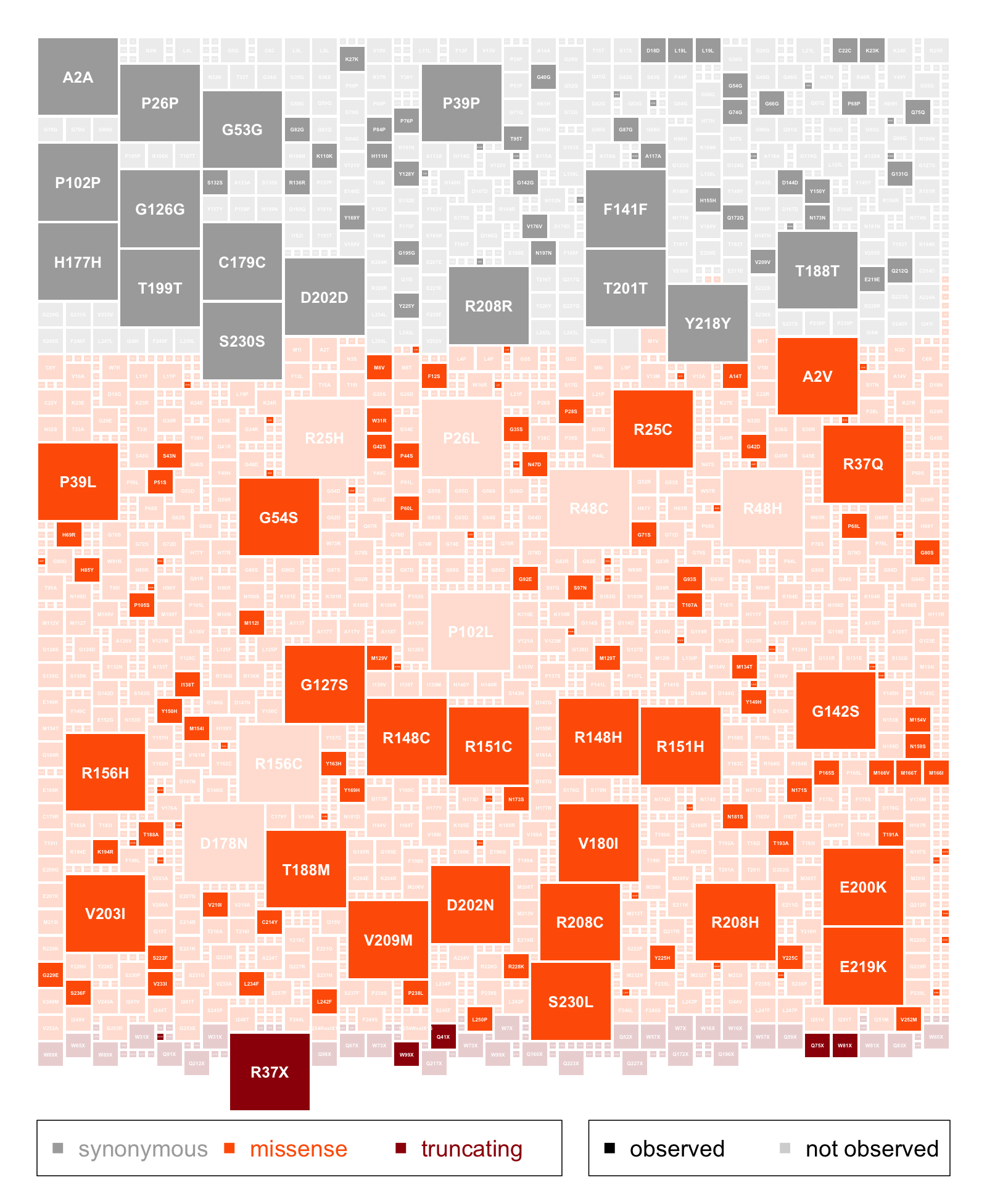
Above: every possible single-nucleotide variant in PRNP, colored by functional class and sized by probability of arising by de novo mutation. Variants seen in gnomAD v2 are shaded at full opacity, while those not seen are at 20% opacity. Click image for PDF version. R code that produced this plot.
Today marks the official publication of the gnomAD v2 package of papers. These seven manuscripts, each in the works for two to five years now, describe what we learned from aggregating the exomes and genomes of a total of 141,456 humans [Karczewski 2020, Minikel 2020, Cummings 2020, Whiffin 2020a, Whiffin 2020b, Collins & Brand 2020, Wang 2020].
I want to take a moment to reflect on a perhaps surprising point: while some of the key insights come from examining the genetic variants we see in these 141,456 people, an equal or greater number of insights come from the variants we don’t see.
The gnomAD studies are in some ways the descendants of a long series of studies cataloguing genetic variation in human populations. First there was HapMap, then 1000 Genomes, then ESP, then ExAC v1, and now gnomAD v2. Over the 15 years of recent history that these studies span, you can see a gradual shift in focus. The early studies were almost entirely about the variants you do see: how common are they, what haplotype blocks do they define, how do they mirror human population structures. Even when HapMap looked at natural selection in human populations, they did so through the lens of the variants you do see: asking, for example, which long haplotypes reflect selective sweeps, and which classes of SNPs are forced by selection to be rarer than others [HapMap 2005, 1000 Genomes 2012].
As far as I know, it was after the advent of 6,503 exomes in ESP that people really started to think about the variants that they didn’t see. (Of course, this may well just be my ignorance: I am not super well read on theoretical genetics, and I am sometimes amazed to discover what elaborate concepts people worked out theoretically decades before molecular biology enabled practical applications.) One approach using ESP data regressed the number of common missense and truncating variants observed against the total number of variants observed, including the synonymous and the rare, in order to derive a “residual variation intolerance score” (RVIS) [Petrovski 2013]. Although this approach relied entirely on counting the variants you do see, a gene’s deviation from the regression line measured how much it was depleted for common functional variants by natural selection. Thus, RVIS indirectly measured what was missing, giving us a first peek into the variants we didn’t see.
That view into the unseen deepened when Kaitlin Samocha formalized a new metric under the name “constraint” [Samocha 2014]. Constraint is based on the number of observed rare variants in a functional class (say, missense) divided by the number expected based on de novo mutation rates. This has conceptual similarities to RVIS, and it does involve regression using the count of synonymous variants in each gene to set expectations. But calculating constraint required, for the first time to my knowledge, not just estimating but actually enumerating the set of all possible variants in a gene. Of course, in principle, the set of possible variants is boundless: make up any DNA sequence of any length, insert it at any point in the genome, and that’s a variant. But if we ignore insertions and deletions and focus on single-nucleotide variants, then the set becomes bounded: about 3 billion positions times 3 possible changes at each, equals 9 billion possible variants. This definition of a set of “possible” variants ends up critically underpinning the ability to learn from what’s not there. I still marvel at the seeming effortlessness with which Kaitlin’s python code, which I first saw in 2014, loops over every codon of a gene, every base of the codon, and every possible mutation. Omitting, for readability, a bunch of error checking and sequencing depth correction details, here is what her loop fundamentally consisted of:
# For each codon
for x in range(0, len(full_context)-1,3):
codon = full_context[x:x+3]
# For each base in the codon
for y in range(x,x+3):
real_loc = context_locations[y]
kmer = full_context[y-1:y+2]
for new_base in ('A', 'C', 'G', 'T'):
if new_base == ref_base:
continue
new_kmer = list(kmer)
new_kmer[1] = new_base
snp_prob = float(snp_probs['{0}->{1}'.format(
kmer, ''.join(new_kmer))])
This enumeration was an important conceptual leap, and missense constraint proved to be a powerful tool for identifying genes, and later regions of genes, where de novo variants were more likely to be disease causing [Samocha 2014, Samocha 2017]. The simplicity and elegance of the above code is due in part, however, to our own ignorance. Our ability to annotate or predict the functional impact of missense variants is so poor that there wasn’t much to do with these variants besides add up their probabilities. So while Kaitlin’s code enumerated every possible variant in each gene’s coding sequence, it did so on the fly, generating and forgetting them on each iteration of the loop.
The sample size in ESP was really just at the threshold where missense constraint could begin to be useful. ESP was nowhere near large enough to start thinking about constraint for protein-truncating variants, for the simple reason that the number of opportunities for new stop codons or essential splice site mutations to arise is far smaller than the number of opportunities for new missense variants. When we began to analyze the data from ExAC v1 — the exomes of 60,706 people — missense constraint had now hit its stride, and truncating variants were now at the threshold where you could just begin to think about constraint. I say “just begin” because, for very long genes, you could get a good estimate of expected number of truncating variants, and show divergence from this, but for most genes of more average length, statistical power was still iffy. Thus, while the underlying model for protein-truncating constraint was still observed divided by expected variants, this was for most analyses abstracted into a metric called “pLI” — probability of being loss-of-function intolerant — that captured both how nominally depleted the gene was for truncating variants, and how confident we were of that depletion [Lek 2016].
While ExAC v1 was early days for protein-truncating constraint, I remember it was around that time that Konrad Karczewski first coined the term “synthetic VCF”. This referred to a totally made-up variant call format file that, unlike most VCFs, did not list variants seen in an individual or population, but instead contained a defined set of all possible variants. Whereas enumeration on the fly had been good enough for missense variants, protein-truncating variants presented a new challenge: lots of them weren’t real. Meaning, some fraction of apparent truncating variants do not, in fact, cause a true loss of function. If you examine the variants you do see, then the more essential a gene is, the more you’ll be looking at artifacts — variants that appear protein-truncating at first glance but do not actually inactivate the protein. To really calculate loss-of-function constraint, therefore it wasn’t enough to just count the set of possible protein-truncating variants, you actually had to store on disk a whole synthetic VCF of all possible variants, and computationally annotate, based on myriad inputs, which ones were likely to cause a true loss of function. This was what Konrad’s LOFTEE — loss-of-function transcript effect estimator — script was designed to do.
Over the past decade-plus, then, our view into the variants we don’t see has evolved several times: from largely ignoring them, to indirectly estimating them, to enumerating them on the fly, to listing them all in a great enormous text file on a cloud server and annotating the heck out of them. In parallel with this evolution, the learnings from each new dataset have skewed more and more towards learning from what we don’t see.
When I look back at ExAC v1, the things we learned seem about equally divided between the “see” and “don’t see” categories. On the “see” side, for instance, we saw supposedly-pathogenic variants in CIRH1A, PRNP, and many other Mendelian disease genes that were more common than they should be, leading us to reclassify them or quantify their penetrance [Lek 2016, Minikel 2016]. We generalized this thought process into an allele frequency filtering model for ruling out potential pathogenic variants based on their observed frequency [Whiffin & Minikel 2017]. On the “don’t see” side, we were able to refine the constraint model from [Samocha 2014] and show how the extent of missing missense variation in a gene correlates with other measures of its essentiality. There were also findings right at the intersection of these two approaches: we realized that we saw a lot of CpG mutations in two different populations, reflecting recurrence — the same genetic variant arising independently more than once in human history. This was an assertion about variants we do see, but, convincing ourselves of how to interpret the data required defining the set of variants we could ever see. The fact that observed CpG mutations had begun to saturate — that we had begun to hit diminishing returns as we neared seeing all possible synonymous CpG variants in the exome, for example — was a key piece of evidence confirming recurrence [Lek 2016].
Today, in the gnomAD v2 analyses, this process of defining the set of possible, and comparing the observed, in order to quantify that which you do not see, has taken center stage. The flagship paper [Karczewski 2020] centers almost entirely on loss-of-function constraint. It lays out the approach to annotating high-confidence loss-of-function variants and defines a loss-of-function constraint metric for human genes — a measure, in essence, of how much you don’t see — and shows how this metric correlates with essentiality in mice and cells, enrichment in disease cohorts, and so on. Another paper [Cummings 2020] shows how to use transcriptomics data to further refine the constraint concept, showing that when a disease-associated gene has some exons that are always expressed and others that are rarely expressed, it is usually the case that the variants you see are in the non-expressed exons, while the burden of missing, don’t-see variants, fall in the expressed exons. A third study provides our first view into constraint in non-protein-coding DNA sequence, showing how 5’UTR variants that would have created upstream open reading frames are depleted in constrained genes [Whiffin 2020b], again quantifying a set of variants that you don’t see because natural selection doesn’t suffer them to exist.
As always, some of the interesting findings in gnomAD v2 are still about variants we do see, such as LRRK2-inactivating variants in healthy people [Whiffin 2020a], and new catalogs and analyses of multinucleotide varaints [Wang 2020] and structural variants observed in gnomAD [Collins & Brand 2020]. Although even then, a lot of the most interesting findings in these papers concern indirect glimpses of what’s missing, such as the classes of non-coding copy number variants that occur less often than expected [Collins & Brand 2020]. All of the things quantified and catalogued by gnomAD — SNPs, multinucleotide variants, structural variants, constraint metrics for each gene — are presented on the gnomAD browser. If ExAC v1 is any guide, probably their single most common practical application will be in clinical genetics, facilitating the interpretation of candidate causal variants found in patients with putative genetic diseases. But we can do more than just diagnose disease: the ultimate goal is to prevent or treat disease, and I believe that this large catalogue of genetic variation also gives us opportunities to think more rationally about drug discovery.
In 2018, Sonia and I announced publicly that we were working on antisense therapy for prion disease with Ionis Pharmaceuticals. Since then, we’ve spoken about our work to dozens of audiences: patients and families, geneticists, medical students, our thesis committees. Without fail, a question we would always get was: are you sure it’s safe to knock down prion protein? Some askers would elaborate: this gene is so important, and we’ve never seen someone without it, so isn’t it scary to go after it with a drug? These are astute questions, and the reproducibility with which they got asked, across diverse audiences, shows how important it is to address them. Over countless evenings of editing our slides to better address these questions up front, I realized there were three conceptual steps people were taking. First, that prion protein stood out above other genes or other potential drug targets in terms of its importance, second, that if it wasn’t so important we’d have seen someone without it, and third, that important genes shouldn’t be targeted with drugs. And I realized that gnomAD data offered us a chance to quantify how all three of these leaps were in fact misconceptions.
Therefore, in my main contribution to the gnomAD research effort, I asked how we should, and shouldn’t, use both the variants we do see, and the variants we don’t see, to inform drug discovery [Minikel 2020].
Addressing the last misconception first, we find that many constrained genes — genes with a lot of “don’t see” truncating variants — are in fact great drug targets. In fact, when we look across targets of approved drugs, we find they are all over the map in terms of how constrained they are against truncating variants. If we limited ourselves to the view that essential or constrained genes could not be drug targets, then we’d have no statins, no NSAIDs. Indeed, about a fifth of all successful, approved drug targets would go out the window. Some of these genes also have lethal mouse knockout phenotypes, and are more conserved in terms of inter-species conservation as well, so the idea that essential genes can be good drug targets has been around — but now we have human data to back it up. This raises the question of why, exactly, some genes seem to tolerate inhibition by a drug but not inactivation at the genetic level. While we can’t offer answers for any specific gene, we can point to a lot of ways in which genetic inactivation is different from pharmacological action — for instance, what tissues are affected, at what age, for what duration of time and to what extent.
Second, moving from the set of “don’t see” variants to “don’t see” genotypes, we model what sample size it would take to find homozygous or compound heterozygous “knockout” humans — people with both copies of a gene inactivated — across human genes. In short, we find that we are not yet anywhere near the sample size where we would expect to find human “knockouts” for a short gene like PRNP, or indeed, for most genes in the human genome. And getting to the sample size where you expect to find a homozygous genotype is one thing; getting to the sample size where the absence of said genotype becomes suspicious is yet a higher bar. In other words, to sequence people and say “we didn’t find homozygous knockouts, therefore that genotype is lethal” is very difficult indeed. For many genes, loss-of-function variants are so rare to begin with that there aren’t enough humans on Earth for us to ever make such a conclusion. At least based on populations with random mating, that is. In people with long stretches of autozygosity — chunks of a chromosome inherited from the same recent ancestor — homozygous genotypes are much more likely, and in such cohorts we may be only a 30-fold increase in sample size away from seeing homozygous loss-of-function genotypes for most genes, as opposed to 1,000-fold.
Third, we show that correctly interpreting that which you “don’t see” sometimes requires deeply curating and correcting for gene-specific biology. For instance, after we account for truncating variants occurring late in PRNP that cause a pathological gain-of-function, we find that PRNP is all the way at the unconstrained end of the spectrum. Compared to virtually any other human gene you could imagine targeting with a drug, we see no evidence that nature dislikes it when people have one copy of PRNP inactivated. Granted, based on the first conclusion above, that constrained genes can be good drug targets, I’d say we should target PRNP even if it were constrained — but the fact that heterozygous loss-of-function seems tolerated gives us an added level of comfort that targeting this gene should be safe. Of course, this isn’t a reason to be cavalier: just like any other novel molecule, PRNP-lowering drugs will have to go through rigorous safety assessments beginning in a small, carefully monitored Phase I trial cohort.
For me personally, the most important take-home here has been how lucky we are to have such a good drug target. But looking genome-wide, at drug targets writ large, I hope the paper provides some useful context for how to discover drugs in light of genomic data — of both the see and don’t-see varieties.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.