The Lancet Summit: presymptomatic prevention and treatment of neurodegenerative diseases
The Lancet Summit: Presymptomatic Prevention and Treatment of Neurodegenerative Diseases was held virtually December 14 - 16, 2021. Due to a busy schedule full of conflicts, I wasn’t able to attend all talks, but those I did attend are blogged below. Note that I confirmed with the conference organizer that the conference’s social media policy allows blogging the content of the talks.
Nick Fox | Overview & Context, Opportunities & Challenges
Dr. Fox gave an introduction framing the importance of the conference’s theme: prevention. He argued that neurodegenerative disease is the challenge of our age, because even as death rates drop for many other disorders including ischemic heart disease (IHD), the death rate for dementia continues to rise. Moreover, because many forms of dementia have long disease duration and a high level of dependency, their cost to society is outsized; dementia is estimated to cost more in the U.K. than heart disease and cancer combined [Winblad 2016]. Society should therefore invest in prevention, in order to lower the prevalence and overall cost of dementia.
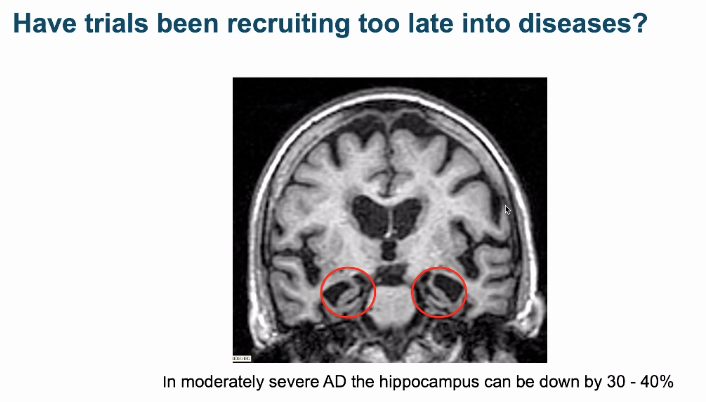
He said it is “not the exception, but the rule” for neurodegenerative diseases to have long presymptomatic stages, with months or decades of disease progression hidden under the surface. He raised the question of whether most therapies tested clinically have been tested too late, and he showed the example of an MRI from an individual enrolled in an Alzheimer’s trial, in whom the hippocampus had already atrophied by 30-40%. How could a therapy be expected to address this damage already done?
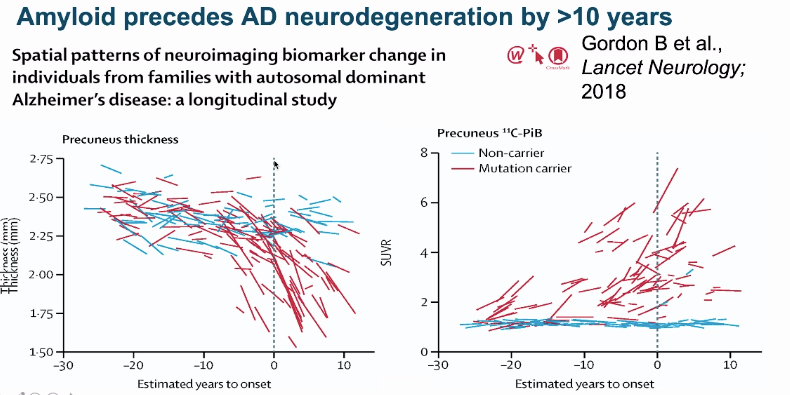
While awareness of prodromal pathology has grown in recent years, he pointed out it’s not entirely new. One early study observed that the age-dependent prevalence of Lewy bodies preceded that of Parkinson’s disease symptoms by decades, and inferred a 30-year prodrome [Gibb & Lees 1988]. But that study relied on brain autopsy data; new tools allow us to track prodromal change in living patients, and give us a new opportunity to intervene early. As one example, he pointed to presymptomatic genetic Alzheimer’s disease, where atrophy of the brain’s precuneus region and accumulation of Aβ plaques occur decades before onset [Gordon 2018]:
What do we mean by “presymptomatic”? For the purposes of this conference, a person might have subtle detectable behavioral changes, but as long as they and their loved ones are not aware or don’t feel anything has changed, they count as presymptomatic. He listed a host of reasons we want to intervene at such a stage, ranging from the molecular — ability to target upstream factors in the disease process, perhaps with a lower dose — to the humanistic — desire to preserve independence and full cognitive function. Nonetheless, he argued that presymptomatic intervention is really challenging, for this list of reasons:

Among other things, overcoming these challenges will require finding new endpoints for trials in presymptomatic individuals, including biomarkers but also perhaps subtle clinical outcomes that are observable before the onset of what we’d call symptoms [Weston 2018].
John Hardy | The genetics of neurodegeneration
Dr. Hardy began by reviewing the Mendelian forms of neurodegenerative diseases. Although copy number variants are very rare, they teach us a very direct lesson. Duplication of the genes encoding the substrate proteins in these diseases — APP, SNCA, MAPT — lead to Mendelian disease, indicating that these proteins are already, in normal people, expressed at a level close to their deposition threshold.
At first glance, GWAS hits for neurodegenerative diseases didn’t always seem to directly relate to the Mendelian hits. For example, Alzheimer GWAS hits are primarily microglial genes, rather than neuronal genes involved in amyloid metabolism. But it turns out that there is a tight mechanistic relationship. TREM2, the first GWAS hit for Alzheimer’s [Guerreiro 2013], turns out to be a key part of a transcriptional program in microglia that responds to amyloid buildup [Huang 2017]. It appears that the unifying theme among many Alzheimer’s GWAS hits is a role in immune response to amyloid deposition [Salih 2019]. Thus, whereas Mendelian forms of AD arise from amyloid overproduction, a lot of the risk for sporadic AD appears to come from defects in amyloid clearance.
Polygenic risk scores (PRS) predict Alzheimer’s pretty well. Using just APOE, you get an area under the curve of 70%; adding all other genomic information gets you to 84% [Escott-Price 2017]. But even monozygotic twins are not perfectly concordant for Alzheimer’s, so it’s unlikely that better PRS are going to do much better than 84%. The PRS breaks down interestingly into stages [Leonenko 2019]. The risk of amyloid deposition is almost entirely driven by APOE, but whether this amyloid deposition progresses to dementia depends additionally on all the other GWAS hits, which are primarily microglial. This divides the process into two stages: first, APOE-dependent amyloid deposition, and second, overwhelming of microglial clearance. In mouse models, while amyloid alone does not lead to tau pathology, by crossing amyloid-overproducing mice with mutant tau mice, it was found that amyloid deposition drives the development of tau pathology [Lewis 2001], Trem2 knockout accelerates this process [Lee 2021] and microglia appear to use TREM2 to manage amyloid and guard against that progression into tau tangles.
Dr. Hardy then shifted gears briefly to Parkinson’s. He said that synuclein in PD is analogous to amyloid in AD, and lysosomes in PD are analogous to microglia in AD. Lewy bodies in synucleinopathies appear to be failed lysosomes that got overwhelmed while trying to clear synuclein aggregates. PD can arise from overproduction of synuclein (SNCA duplication) or from defects in lysosomal clearance (GBA mutations).
A unifying view of many/most neurodegenerative diseases is that each can arise from either an overproduction of a substrate, or from an age-dependent failure of clearance mechanisms. But how does amyloid lead to tau pathology in AD? An outdated view is that Aβ triggers tau misfolding through, say, a receptor or something [Hardy 1998]. But it’s not just tau — many AD patients also have other co-pathologies such as Lewy bodies. Therefore, Dr. Hardy now believes that amyloid simply overwhelms protein clearance mechanisms including both lysosomes and proteasomes, and once these systems are overwhelmed, all different pathological proteins can start to build up, of which tau is just one.
The therapeutic strategies these hypotheses imply are 1) lower substrates, for instance through ASOs, or 2) enhance clearance, for instance through antibodies.
Virginia Lee | Transmission of misfolded proteins in neurodegenerative disorders: potential approaches in the next decade
Dr. Lee introduced protein aggregation as a common feature across several different neurodegenerative diseases. In many cases, the aggregation spreads in a stereotypical regional pattern from a particular starting point [Jucker & Walker 2013]. As of a decade ago, two core questions were outstanding in many of these diseases: are they transmissible, and do the misfolded proteins come in different strains that explain phenotypic variability? Dr. Lee’s lab established pre-formed fibrils (PFFs) of alpha-synuclein injected into wild-type mice as a model for synucleinopathies, confirming the fundamentally transmissible nature of this pathology [Luk 2012]. However, to their disappointment, when they made heparin-induced aggregates of Tau40 (Hep-T40 PFFs), these did not induce any significant tau pathology. It turned out that tau aggregates from Alzheimer’s brain, however, had unique conformational properties and were able to induce pathology in wild-type mice [Guo 2016], thus validating both transmissibility and strains in tau. The propagation of tau pathology in these injected mice followed the neuroanatomical connectome. Further in vivo work showed that tau strain diversity can account for phenotypic diversity [Narasimhan 2017], For TDP-43, they found that conditional expression of TDP-43 with a defective nuclear localization signal gave rise to some limited pathology [Igaz 2011], but if they injected them with TDP-43 extracts from FTLD patient brains, they got robust pathology [Porta 2018].
Dr. Lee posited that the validation of transmission in neurodegenerative disease protein aggregates gives rise to four therapeutic hypotheses for each protein: block release of aggregates from diseased cells, promote degradation outside the cell, block uptake into healthy cells, or prevent seeding in the healthy cell. This is in addition to other strategies like lowering the expression of the substrate protein. As an example, she pointed to recent work showing that TREM2 is involved in controlling the spread of tau pathology [Leyns 2019], which might be mediated by degradation of aggregates.
Not all therapeutic hypotheses are relevant at all phases of disease: for example, in Alzheimer’s, it has been posited that therapies targeting Aβ might be effective only decades before onset, with tau therapies becoming relevant at later presymptomatic stages, whereas at the symptomatic stage where most clinical trials take place today, it may be too late for either approach, and symptom management may actually be the only strategy remaining.
Ammar Al-Chalabi | An overview of ALS genetics in general with discussion of prevention
Dr. Al-Chalabi drew a distinction between “familial” and “genetic”. ALS is common enough (~1 in 300 people) that occasionally there will be more than 1 case in a family without a mutation, especially in large families [Byrne 2011]. Conversely, penetrance is <100%, so there will occasionally be genetic families with only one person affected, especially in small families [Al-Chalabi & Lewis 2011]. Further complicating matters, “sporadic” ALS has some heritability, so family members of ALS cases without a mutation are still at some increased risk [Hanby 2011]. When they asked geneticists to look at eight pedigree diagrams and decide if they were “familial” disease or not, there was a spectrum of opinions on which pedigrees had cases densely clustered enough to count as “familial” [Byrne 2012]. There is also a potential for ascertainment bias that could hypothetically lead to a younger observed age of onset in cases that are part of familial clusters, however, it turns out that the age difference between “familial” and “sporadic” ALS is explained entirely by whether you have a mutation [Mehta 2019]. Dozens of Mendelian ALS genes have been discovered, with the number doubling every four years [Brown & Al-Chalabi 2017]. There are still many variants of unknown significance in these genes, which is a challenge for clinical trials, as if someone is enrolling in a trial of a therapy targeting a particular gene, you’d better know that their ALS is caused by that gene and not an idiopathic case coincidental to a benign rare variant in that gene. There is significant shared genetic risk from common variants between ALS and other neurodegenerative diseases, suggesting shared pathways [van Rheenen 2021].
Where is the opportunity for primary prevention? ALS is rare enough that any advice to reduce risk can’t be given to the general population if it conflicts with general health advice. For instance, you wouldn’t tell people to exercise less, even if that reduced ALS risk. Advice or interventions to reduce ALS risk are only relevant to people with heightened risk of ALS. How do you find those people? Genetic ALS is too rare for general population screening to be helpful — the test would have to be virtually perfect — but we can screen people with family history. There is some Mendelian randomization evidence that increased total cholesterol contributes to ALS risk [van Rheenen 2021]. Likewise, Mendelian randomization also found that people with genetic predisposition to exercise a lot also had higher risk of ALS [Julian 2021], though this doesn’t necessarily indicate that exercise itself increaases ALS risk. The increase in ALS risk with age as well as with genetic mutations can be combined in a sort of “multistep” model explaining ALS onset [Chio 2018].
Dr. Al-Chalabi used the term “secondary prevention” to refer to preventing further progression in symptomatic patients. He speculated this might need to be done in a genotype-targeted manner, citing meta-analysis evidence that ALS patients may respond differently to lithium depending on their genotype [van Eijk 2017].
In Q&A, there was a lengthy discussion about pathways involved in ALS. Dr. Al-Chalabi noted that many ALS risk genes converge on TDP-43 pathology, but that it appears SOD1 and FUS do not. He noted, though, that for therapies aimed at lowering expression of the causal gene and tested in a population with mutations in that gene, the pathway may not matter. Dr. Tabrizi, hosting the session, asked by what mechanism exercise, or genetic predisposition to exercise, might contribute to risk. Dr. Al-Chalabi discussed several theories but that it’s not really known. Dr. Tabrizi asked what the mechanistic hypothesis was that led to lithium being tested in ALS trials, and Dr. Al-Chalabi replied that there hadn’t originally been a strong scientific foundation for hypothesizing that lithium could be helpful.
Reisa Sperling | Alzheimer’s
Dr. Sperling began by reviewing the rationale for biomarked-based preventive trials in Alzheimer’s. Among cognitively normal older adults, brain amyloid accumulation is associated with subsequent cognitive decline [Donohue 2017] as well as tau pathology, which particularly accelerates at some particular threshold of amyloid [Sanchez 2021]. Dr. Sperling defined the following terms:
- Primary prevention: preventing people from developing amyloid or tau pathology in the first place
- Secondary prevention: finding people with amyloid or tau pathology and preventing progression to cognitive impairment
Dr. Sperling discussed the A4 trial [Sperling 2014, Insel 2020], a secondary prevention trial in which people with amyloid pathology are randomized to an Aβ antibody solanezumab, or placebo. It is powered for a 30% decrease in rate of cognitive decline in the treated group. They had to screen 50,000 individuals by web or phone to find 6,700 that merited in-person screening, which led to florbetapir amyloid PET imaging on 4,468 individuals, of whom 1,169 with brain amyloid were randomized, with enrollment completed in December 2017. Even though all these people screened as being in a very tight range of normal cognition, several subtle measures of cognition and self-reported function were slightly but significantly altered in the Aβ positive group vs. Aβ-negative people who were screened out [Sperling 2020, Amariglio 2021]. They also see evidence of tau accumulation in this group. All these findings made Dr. Sperling group, what if even the A4 trial is too late — what if they’re targeting Aβ after Aβ has already led to tau pathology and cognitive decline? Could we go even earlier instead?
Dr. Sperling more recently has launcehd the AHEAD study, which includes the A3 and A45 randomized studies. Where A3 tests the ability to prevent Aβ accumulation in amyloid-negative, “pre-preclinical” people, while A45 tests intervention in people with a higher amyloid burden than A4. This trial is a public/private partnership with NIH ACTC and Eisai, with dosing of lecanemab, another Aβ antibody.
In Q&A, someone asked how touch it was to recruit people for these trials. Dr. Sperling said there was actually a waiting list for A4 — there was a tremendous amount of interest. The problem was the efficiency of screening, with only about 2% yield.
Sarah Tabrizi | Huntington’s disease and HTT lowering genetic therapy trials
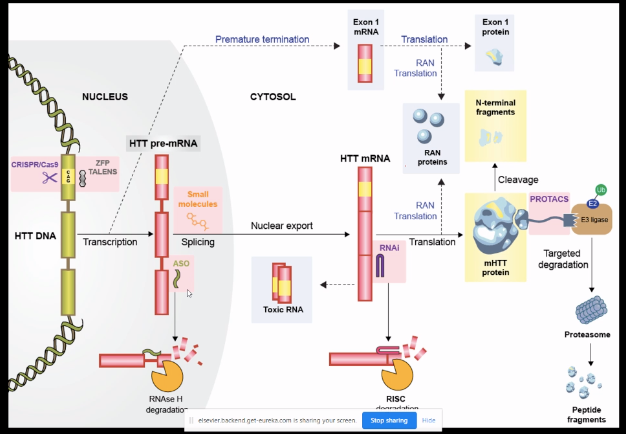
Dr. Tabrizi showed a diagram of all the different strategies currently in development for HTT lowering:
Dr. Tabrizi described in detail the results so far of HTT-lowering ASO trials, which I’ve blogged here. While she wasn’t able to share any new data beyond what Dr. Scott Schobel shared back in April, she offered her opinion on possible reasons the trial failed:


In particular, she pointed out that the disease stage in GENHD1 was likely too late and that we need earlier intervention — the topic of this whole conference.
What’s next now that tominersen has failed? She reviewed a few ongoing efforts including:
- allele-specific ASO WVE-03 from Wave, which I’ve covered here
- AAV5-vectored HTT exon 1 miRNA from UniQure, for which preclinical data have recently been published [Valles 2021]
- splice modulators branaplam and PTC-581, which introduce a pseudoexon in HTT mRNA between exons 49 and 50 resulting in nonsense-mediated decay. Both of these small molecules are already in Phase I and have Phase II trials in early or prodromal HD launching in the next few months.
Host Dr. John Hardy asked whether the negative result from tominersen has diminished enthusiasm for trial participation in the HD community. Dr. Tabrizi said that enthusiasm appears undiminished.
Feng Zhang | Harnessing biological diversity for developing programmable therapeutics
Dr. Zhang divided his talk into three stories.
First up, the exploration of diversity of naturally occurring CRISPR-Cas systems to expand the genome-editing toolbox. Class 1 systems use a multi-subunit crRNA-effector complex. Class 2, including Cas9, use a single-subunit complex, which is more tractable for genome engineering and therapeutics. They therefore sought to find other Class 2 systems besides Cas9 [Shmakov 2015]. They found a few different versions of Cas12, which are DNA-targeting, and a few different forms of Cas13, which are RNA-targeting, since bacteria also need to defend against attack by RNA viruses. Interestingly, there turn out to be systems with Tn7-like transposons that lack DNA recognition capability, combined with CRISPR-Cas systems that lack nuclease activity [Peters 2017]. It turns out these combinations use CRISPR to recognize a site for the transposon to insert in the genome — RNA-guided DNA insertion [Strecker 2019].
Second, developing non-viral delivery technologies. One of the great limitations of any sort of CRISPR system is how to deliver it to disease-relevant cells. Some retroviruses got into the human germline many eons ago, and some retroviral genes even took on important native functions in human biology. One retroviral protein called ARC is expressed in the brain and forms a virus-like structure, which made them wonder if things like this could be hijacked for drug delivery. After screening a panel of mammalian retrovirus genes, they found that the PEG10 gene encodes a protein that forms a virus-like particle that packages its own RNA. By putting the PEG10 untranslated regions on to a cargo gene of interest, one can get a cargo gene packaged into PEG10 protein and delivered into cells [Segel 2021]. This is a new potential method for non-viral delivery of gene therapy.
Third, genome editing capabilities beyond CRISPR-Cas9. By examining the phylogeny of the most conserved elements of Cas systems, they found distant relatives IscB, IsrB, and TnpB, which are RNA-guided DNA nucleases that are analogous to CRISPR-Cas9 but do not have a CRISPR element and instead have a different sort of RNA guide [Altae-Tran & Kannan 2021]. IscB is present across various domains of life (including algae) and not just in bacteria. Like CRISPR, they were able to reprogram this system to target a DNA sequence of interest, establishing a potential as a tool for use in research and in therapeutics.
Roy Yaari | Eli Lilly’s preclinical Alzheimer’s disease program
In Dr. Yaari’s title, the term “preclincial” means “pre-symptomatic”, as opposed to “animal”. He structured his talk around two pre-symptomatic Alzheimer’s trials.
The first half was about solanezumab, an Aβ antibody. Solanezumab has been tested extensively at a low dose (400 mg every 4 weeks) in symptomatic AD, with no positive results, but a trend towards positive outcomes on a few measures in the earliest/mildest patient groups. They then launched a trial specifically in very early/mild AD and again saw a positive trend but still not significant. This inspired two changes: an escalation of dose to 1600 mg every 4 weeks, and a move to earlier patients in the “anti-amyloid treatment in asymptomatic Alzheimers” (A4) trial. See Dr. Sperling’s talk below for more info about A4. The primary outcome is a cognitive measure. Results will come out in 2023.
The second half of the talk was about donanemab, also an Aβ antibody. The Phase I and II trials all looked very promising in terms of target engagement, and indeed, in early symptomatic patients, there was a nominally significant (P < 0.04) slowing of cognitive decline on the primary endpoint of iADRS score [Mintun 2021], although most secondary outcomes showed no difference. Again, post-hoc analyses suggested a greater benefit for patients treated earlier. This inspired the TRAILBLAZER-ALZ 3 study, which aims for 3,300 cognitively unimpaired patients with biomarker evidence of AD pathology based on plasma p-Tau. Almost all trial activities are conducted remotely rather than with in-person study visits, which they hope will decrease burden on study sites as well as allow recruiting a larger number of people and from more diverse populations. The primary outcome is a time-to-event, where the event is to have two consecutive CDR tests with a score >0, meaning the patient has become cognitively impaired. The trial will stop when they hit 434 events.
Holly Kordasiewicz | Antisense oligonucleotides for the treatment of neurodegenerative disease
Dr. Kordasiewicz reviewed the clinical experience to date with intrathecal ASOs for neurological diseases, spanning one approved drug (nusinersen for SMA) as well as 10 other ASOs that have reached first-in-human:
There are now clinical data from humans treated with three different RNAse H-mediated intrathecal ASOs, including new data just released for MAPT, all showing that ASOs engage their targets in the CNS:
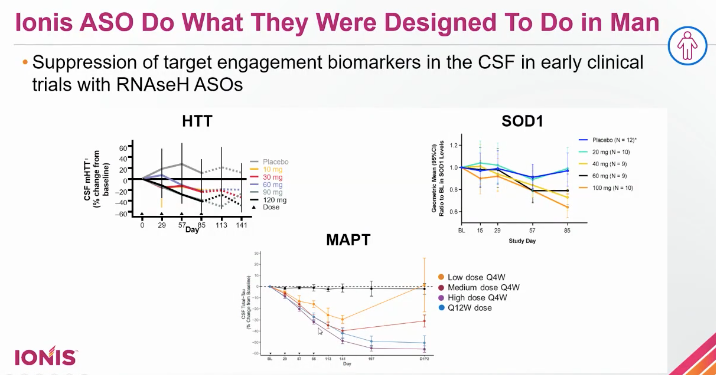
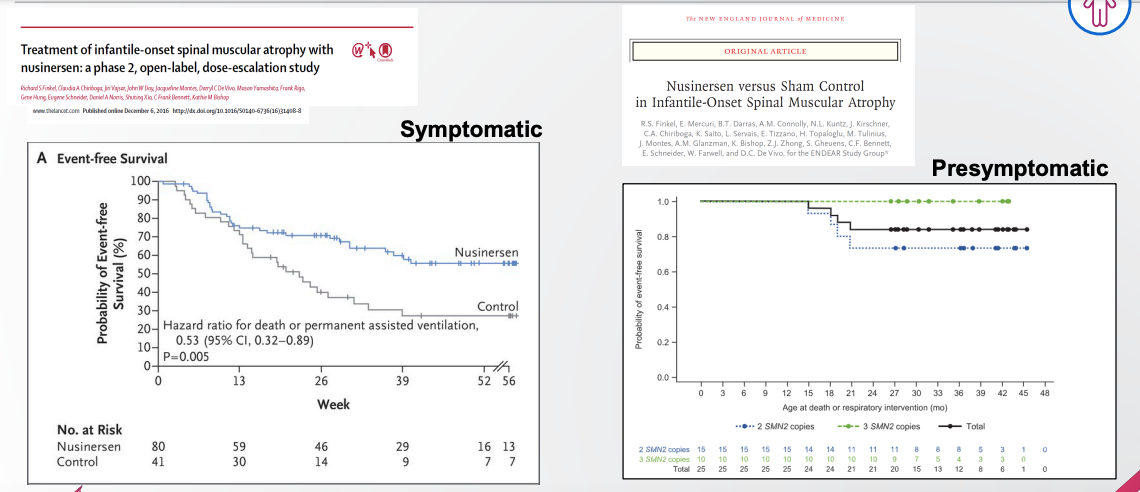
Across multiple programs, she pointed out, there is evidence that earlier intervention is better than later intervention. For spinal muscular atrophy (SMA), the ENDEAR study in presymptomatic infants (right) [Finkel 2017] showed that nusinersen resulted in better progression-free survival than was observed in symptomatic infants (left) [Finkel 2016]:
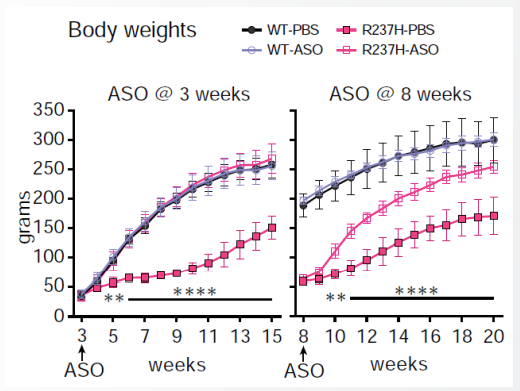
A similar trend has been observed in animal models for a number of ASOs that are now in earlier stages of clinical development. Dr. Kordasiewicz pointed to the drug candidate ION373, targeting GFAP for Alexander disease, which is now in Phase I [NCT04849741]. The evidence from a rat model suggests a superior benefit when treatment is initiated presymptomatically [Hagemann 2021]. Across a number of measures, treated rats were able to fully recover, but the recovery was swift when they were treated early whereas it was slow when they were treated late. In terms of weight gain, a measure of healthy development, rats treated at 3 weeks (presymptomatic) were fully rescued, with weight gain indistinguishable from normal rats, while rats treated at 8 weeks (symptomatic) were only partially rescued, with weight gain about halfway between untreated rats and normal rats:
In mouse models of Huntington’s disease, lowering HTT earlier rather than later did more good [Kordasiewicz 2012]. In our work on prion disease, early chronic treatment with a PRNP-lowering ASO tripled survival, while late treatment only increased survival by 20% [Minikel 2020].
The ATLAS trial in presymptomatic SOD1 mutation carriers may provide the first glimpse of whether this outsize benefit to early intervention replicates in humans.
Robert Klitzman | Presymptomatic prevention trials: ethical considerations
Dr. Klitzman is the director of the Master’s in Bioethics program at Columbia University. He reiterated the basic bioethical principles of respect for persons (autonomy), benificence, non-maleficence, and justice, but noted that interpretation and application can vary, and thus the question of how they apply to preventive trials merits a special discussion.
As for “respect for persons”, informed consent is critical as always. Cognitive impairment in understanding the consent, and risks of stigma or discrimination in genetic testing are special issues in this area. Another issue is “therapeutic misconception”, which is a patient’s belief that because the doctor is in a white coat, they’re doing the best thing for the patient, when in fact it’s research and the patient may not directly/individually benefit; alternatively it is phrased as “denial of the possibility that there could be disadvantages to participating in research”. The possibility of incidental findings, and whether they’ll be disclosed, should be addressed up front. The U.S. has only about 3,000 genetic counselors — one per million people — and many studies may find it hard to staff enough of them to counsel everyone if they’re really going to return incidental findings.
As for “beneficence”, in preventive trials, only people selected to be at high risk should be included, since otherwise some people who would have never become sick could experience risks without any prospect of benefit.
In the discussion panel, Dr. Cristina Sampaio asked Dr. Klitzman about the tradeoffs inherent in legislation designed to protect patient privacy. In Europe, GDPR seeks to protect patients from exploitation of their data, but it is so restrictive that patients are frustrated that it’s not possible for them to contribute their data to research in the manner they would like to. Dr. Klitzman replied that this is an important area for patient advocates to engage with policymakers and try to change it. Meanwhile, there may be workarounds, whereby if Country A doesn’t allow data sharing but Country B does, the sample could be collected and submitted for research in Country B. Another workaround relies on differential regulations for sharing of biospecimens vs. data — for instance, sometimes even if DNA can’t be sent across borders, the DNA could be sequenced in the originating country and the data could be sent across borders.
Lorenzo Guizzaro | Innovative trial design and getting drugs approved in Europe and the UK: regulatory perspective
Dr. Guizzaro prefaced his presentation by saying that this will be his personal opinion and does not represent the official position of the European Medicines Agency (EMA) for which he works; refer to EMA guidance for official positions.
Dr. Guizzaro described the steps for designing a preventive trial as follows:
- Define a population of interest. This could involve selection on a biomarker value, either through single measurements or serial measurement. Before you can use a biomarker as a selection criterion, you need to characterize its prognostic value for predicting a clinical outcome of interest. This can be done through retrospective studies but with caveats, such as availability of historical measurements, ascertainment bias, and so on. At EMA, candidate biomarkers that may be used for patient selection (or as endpoints) are expected to go through a Qualification Procedure. Academics working on qualifying a biomarker can seek early engagement with EMA through the Innovation Task Force.
- Demonstrate clinically relevant efficacy. Unfortunately, Dr. Guizzaro said, this typically is done through a randomized trial design. Exceptions to this need to be justified on knowledge of A) the pathological basis of disease and B) natural history of the disease, such that a non-randomized trial can be deemed to provide equally high quality of evidence as a randomized trial would. The endpoint could be a surrogate biomarker if it is validated appropriately. But we cannot afford a “naive view” of surrogate biomarkers. The “naive view” assumes that because a biomarker is prognostic of a clinical outcome, pharmacologic modulation of that biomarker must therefore predict modification of that clinical outcome. This is naive because it assumes that temporal relationships observed in longitudinal studies are causal relationships. In fact, an alternative interpretation for many biomarkers is that an underlying pathological process causes both the biomarker change and the clinical outcome, accounting for their correlation in longitudinal studies without implying that all interventions which modulate the biomarker modulate the clinical outcome. Another interpretation is that there is some upstream pathological process that triggers the biomarker change, but also triggers a downstream pathological process that then independently leads to the clinical outcome, so that an intevention could target the upstream process and thus change the biomarker, without affecting the downstream process and the clinical outcome.
- Balance risks and benefits in the target population. Depending on the positive predictive value of the biomarker used for patient selection, it might be the case that the intervention benefits only a subset of the population intended for treatment; the remainder would only be exposed to the risks of intervention without any prospect of benefit. This will be taken into account in how regulators assess the risk/benefit balance. Sometimes, extrapolation of safety and efficacy data from a symptomatic population can help corroborate findings. This would be relevant if a drug were already approved for symptomatic treatment and the goal were to expand its use into a presymptomatic population. But this is not helpful in cases where a drug might only work at a presymptomatic stage.
Dr. Guizzaro concluded by discussing the example of nusinersen for spinal muscular atrophy. He pointed to EMA’s Assessment Report (21 April 2017) on nusinersen as a great resource for understanding the agency’s thinking on this drug. He described the evidence from presymptomatic infants as being “very limited” — only 13 subjects — but the results in that small cohort were very good, and there were supportive safety and efficacy data from symptomatic infants. Therefore EMA felt that risks and benefits were balanced, and it approved nusinersen for both presymptomatic and symptomatic treatment.
In the discussion panel, Dr. Cristina Sampaio described Dr. Guizzaro as “throwing cold water” on the enthusiasm for preventive trials, which Dr. Guizzaro objected to. He said that just as we did with FDA, folks seeking preventive clinical paths should engage early with EMA.
Dr. Sampaio also drew a distinction between loss-of-function disorders like SMA, where replacing even a small amount of the missing protein can yield a robust clinical benefit, and where extrapolation from symptomatic to presymptomatic stage is easy. In contrast, she said, in gain-of-function diseases like HD there might simply not be an opportunity for benefit in the symptomatic phase. Thus, the type of “extrapolation” Dr. Guizzaro referred to may not be helpful in HD, AD, or prion disease, and she suggested that the regulatory framework needs to change. Dr. Guizzaro replied that a new framework is not needed — the current framework actually is capable of handling the complex case of HD and other slowly progressive gain-of-function disorders. It’s just that a large body of basic research is needed to convince regulators that the biomarker is strong enough to stand in for a clinical endpoint.
Dr. Sampaio further opined that the EMA requirements for qualification of surrogate biomarkers are virtually impossible to meet without already having a successful clinical trial that moves a clinical endpoint, in which case the surrogate biomarker pathway is only helpful for expanding indications and not for developing first-in-class drugs that we so desperately need. She said the U.S. solution has been the “reasonably likely” language that FDA uses, which is arguably too subjective but is better than EMA’s approach which may be too restrictive. Dr. Guizzaro replied that in EMA, not so different from FDA, it comes down to the regulator’s degree of certainty as to the relevance of the biomarker to the clincal outcome. In establishing that degree of certainty, all lines of evidence you can imagine do come into play — natural history, molecular understanding of the disease, animal models, and so on. Early engagement with regulators is critical, as regulators will be more conservative if approached with final data than if they were involved in designing the biomarker qualification process from day one. Dr. Sampaio agreed that a true partnership is crucial.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.