STX6: validating and invalidating a drug target in the same breath
Here’s a study I’ve been waiting to see for a while: prion infection of Stx6 knockout mice [Jones & Hill 2023]. To me, it beautifully illustrates the challenges in trying to go from GWAS hit to validated drug target.
The backstory is that 3 years ago, the largest-ever genome-wide association study (GWAS) in sporadic prion disease found, for the first time, two significant hits at loci other than PRNP [Jones 2020]. One linkage peak included two missense variants in GAL3ST1 with an unknown direction of effect. The other mapped to an eQTL in STX6, where the allele associated with higher expression was associated with higher disease risk. This implied that pharmacologic inhibition of syntaxin-6 could potentially be beneficial. Fortuitously, the exact same higher-expressing haplotype was also a risk allele in a tauopathy, providing a tantalizing opportunity for a genetically validated target shared between two different neurodegenerative diseases.
The natural next step, reported in the new study, was to infect Stx6 knockout mice with prions and see whether their genotype affected their incubation time. Long story short:
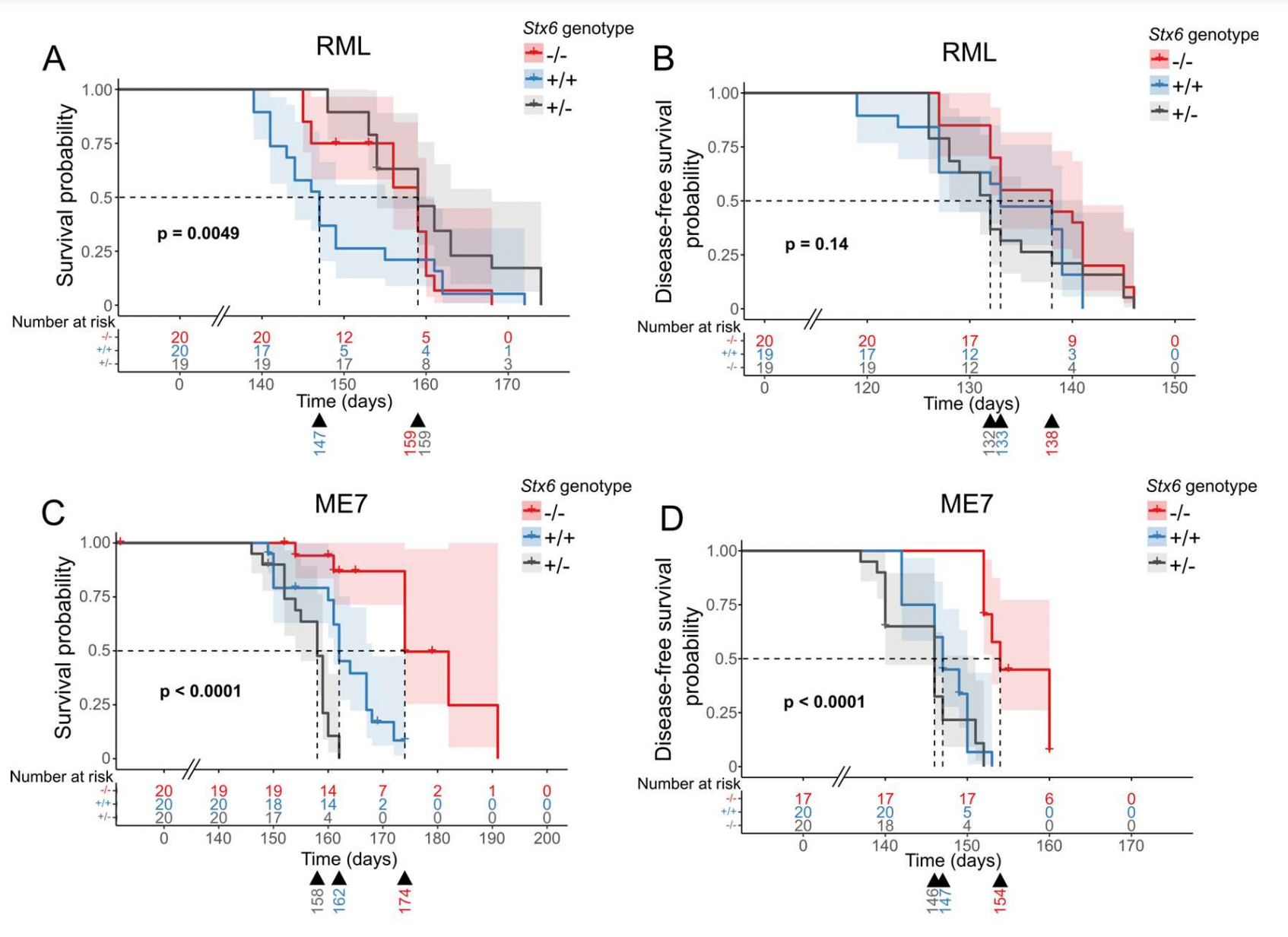
Against two different prion strains, RML and ME7, Stx6 knockout brought about a statistically significant but extremely small delay in disease endpoint — about 12 days (note the broken axis). As a rule, I generally don’t let myself believe small effect sizes in mouse survival curves reported in the prion literature. But here, you have both a stronger prior than usual, since the gene is a GWAS hit, as well as slightly better evidence than usual, with independent experiments in two prion strains. On balance, I tend to believe that this effect could well be real. In that sense, this study validates STX6 as a potential drug target.
At the same time, the small effect size effectively invalidates it too. If even total, lifelong knockout of syntaxin-6 yields only a 12-day difference in disease tempo, what hope is there for late, incomplete, transient pharmacological inhibition to make enough of a difference that one ever could power a clinical trial in symptomatic patients? To be clear, for a disease with no standard of care, anything, even a small therapeutic benefit would be welcome — but it’s hard to imagine anyone wanting to undertake the massive investment to develop a drug against syntaxin-6 for a benefit that might be too small to ever demonstrate. The original GWAS hit at STX6 likewise had only a small effect on sCJD risk (an odds ratio of 1.16), but that was for an eQTL that had but a modest effect on STX6 expression. The hope was that more potent modulation of syntaxin-6 function would yield more potent modification of disease. Apparently not.
Alternatively, we can also consider the pre-symptomatic community. It’s possible — likely, even — that syntaxin-6 genotype has a larger effect on prion initiation than it does on prion replication. After all, it was originally a GWAS hit for sporadic CJD risk, in a case/control design, and did not appear to modify disease duration [Jones 2020]. But trying to develop a syntaxin-6 inhibitor for pre-symptomatic patients would be hard for all sorts of reasons. Syntaxin-6 knockdown/knockout does not affect PrP expression [Jones 2020, Jones & Hill 2023], and indeed, the new paper shows that it doesn’t have very much impact on prion neuropathology either [Jones & Hill 2023]. So a biomarker-based pathway in pre-symptomatics does not seem likely. Indeed, even trying to validate in a mouse model the effect of syntaxin-6 on prion initiation would be rather hard. As we learned the hard way, finding cold hard disease endpoints in a genetic prion disease mouse model can be tough [Vallabh 2023], and I worry that if you use a PrP-overexpressing model to drive a faster, more robust disease outcome, you could miss your chance to modify that outcome by knocking out a different risk gene.
My take, then, is that this is probably the end of the road for syntaxin-6 as a drug target in prion disease. We’ll see if anyone ends up developing it for tauopathies instead.
It’s one more interesting example of how you never know what you’ll learn when you go back to functionally validate a GWAS hit. We know that GWAS hits can make successful targets despite their small effect sizes [Nelson 2015], but again, the hope is always that a drug is more potent than the original genetic variant, and thereby yields more impact on disease. The classic example is that a SNP in HMGCR only affects LDL cholesterol by 8% and coronary artery disease risk by 8%, but statins, which inhibit HMGCR’s protein product, can lower LDL by 19-37% and lower coronary heart disease-related deaths by 23% [Wilt 2004]. But then there are examples like GPR151 where GWAS found that heterozygous loss-of-function modestly reduces risk diabetes and body mass index, but a subsequent deep dive found that the effect of total knockout was null or maybe even paradoxically increased risk [Gurtan 2022]. When you see a new GWAS hit, you don’t know if you’ve just hit upon an HMGCR or a GPR151. But it’s very much worth finding out, and I’m glad that they did this follow-up study on STX6. I wonder, is anyone doing GAL3ST1, the other new GWAS hit? For the time being, PRNP continues its long reign as the sole drug target in prion disease.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.