Evidence that the effect of C-terminal truncating variants in PRNP may be position-dependent
Last year I blogged about PRNP in gnomAD v4, which now contains DNA from 807,192 people, and with it, possibly some new insights about prion biology. One surprise was that there were more protein-truncating variants (PTVs) at the C-terminus of PRNP than we expected. This led me to hypothesize their effects might be position-dependent. This blog post is a deeper dive on that question.
background
Since 2016, the evidence from ExAC and then gnomAD v2 had supported a model where N-terminal PTVs, up through codon 131, were benign loss-of-function variants, while C-terminal PTVs, from codon 145 onward, were pathogenic gain-of-function variants [Minikel 2016, Minikel 2020]. N-terminal ones appeared in gnomAD as often as expected based on mutation rates without natural selection, while the C-terminal ones were depleted fron gnomAD and enriched in prion disease cases, consistent with their being pathogenic. Whereas “typical” prion disease is a rapidly progressive dementia that kills patients in half a year, C-terminal truncating variants in PRNP cause an “atypical” phenotype where many patients present first with chronic diarrhea due to PrP amyloid accumulation in the GI tract, leading to misdiagnosis with inflammatory bowel disease (IBD); over decades they eventually progress to peripheral neuropathy and then a slowly progressing dementia easily misdiagnosed as Alzheimer’s [Mead & Reilly 2015].
This type of atypical prion disease is very rare: prion disease causes ~1 in 6,239 deaths [Maddox 2020], but C-terminal PTVs only account for 0.06% of cases (~15% of cases are genetic [Minikel 2016] but most are due to missense variants). Based on disease prevalence, then, out of 807,192 individuals in gnomAD v4, we would not even expect to see 1 individual with a pathogenic C-terminal PTV by chance. But in gnomAD v4, we saw 15 C-terminal PTV alleles (at or after codon 145) versus ~0.07 alleles on expectation. This was very surprising.
In that 2024 post, I argued there were 2 possible explanations:
- Phenotypic enrichment. gnomAD v4 might contain disease cohorts that enriched for phenotypes of PTV-related prion disease, such as dementia, or inflammatory bowel disease.
- Positional effects. Maybe not all C-terminal PTVs are pathogenic, and the tolerated ones appear in gnomAD.
To explain more the 2nd hypothesis, here’s a zoom in on the C-terminus of PRNP showing where PTVs seen in prion disease cases and in gnomAD (v4 plus earlier versions) occur relative to secondary structure elements of the protein. Note that this is updated compared to the 2024 post, as here I’ve now properly accounted for the fact that a couple of variants (Q186X and Q226X) are seen in both cohorts:
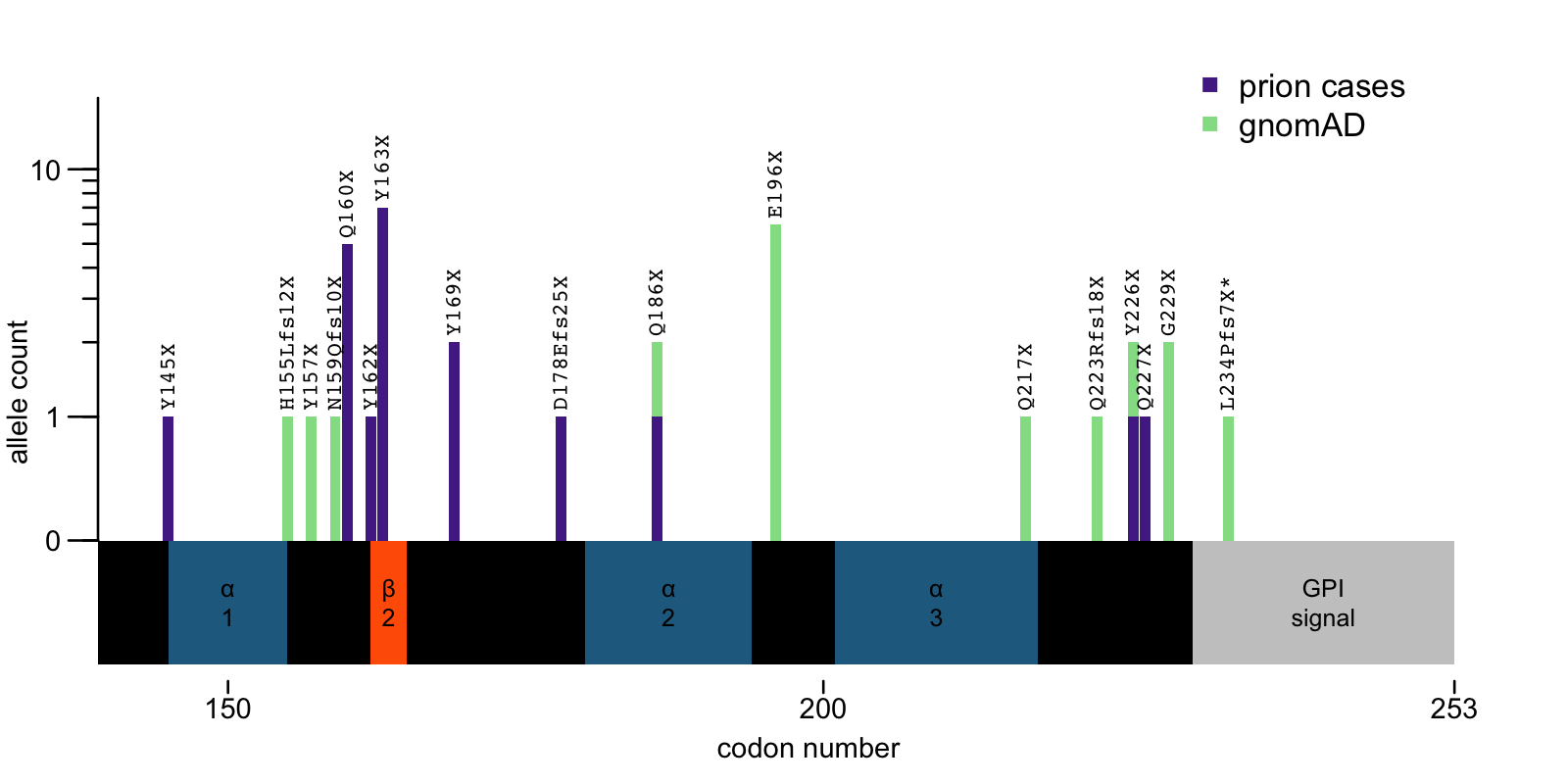
Above: C-terminal PTVs in PRNP. *L234Pfs7X was seen in a gnomAD dementia cohort; the patient had a diagnosis of Alzheimer’s but was not autopsy-confirmed, see [Minikel 2020].
Now the question becomes, are those new individuals from gnomAD v4 from dementia or IBD cohorts?
Genebass
Genebass is a public browser of exome burden test results for phenotypes in UK Biobank samples, and can be used to glean some information about phenotypes associated with individual variants of interest. When I enter PRNP versus the phenotype “FH Alzheimer disease dementia custom”, test for burden set pLoF, and filter to show only the PTVs, here is what I get:
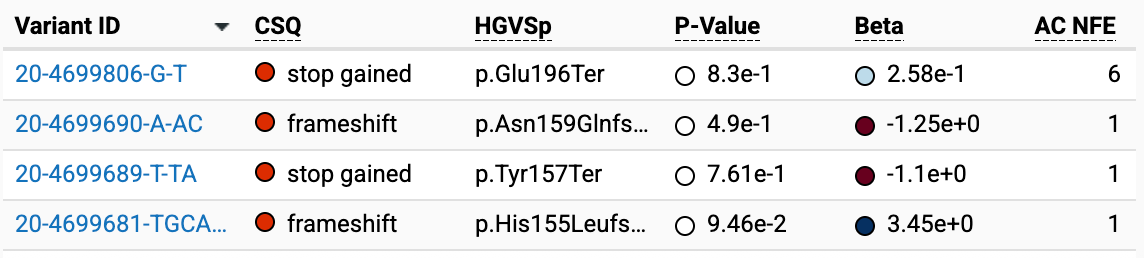
In other words, of the C-terminal PTVs we are interested in, 4 unique variants and 9 alleles are from UK Biobank.
For variants with AC=1, a positive beta means that the one individual does have the phenotype, and a negative beta means they do not. Thus: H155Lfs12X is a “case” (in this case, the phenotype is “FH Alzheimer disease dementia” so probably someone with family history (FH) of Alzheimer’s), while Y157X and N159Qfs10X are in controls.
E196X has an allele count of 6 so it is a slightly tougher nut to crack. The phenotype “FH Alzheimer disease dementia custom” has 53734 cases and 327258 controls. We know that the beta is 0.258 and the P value is 0.83. The number of E196X individuals that are cases could be 0, 1, 2, 3, 4, 5, or 6. While the burden test isn’t exactly a Fisher exact test, it’s similar, and we can get some idea by trying every combination:
cases = 53734
controls = 327258
fisher.test(matrix(c(6, cases-6, 0, controls-0), nrow=2, byrow=T))
fisher.test(matrix(c(5, cases-5, 1, controls-1), nrow=2, byrow=T))
fisher.test(matrix(c(4, cases-4, 2, controls-2), nrow=2, byrow=T))
fisher.test(matrix(c(3, cases-3, 3, controls-3), nrow=2, byrow=T))
fisher.test(matrix(c(2, cases-2, 4, controls-4), nrow=2, byrow=T))
fisher.test(matrix(c(1, cases-1, 5, controls-5), nrow=2, byrow=T))
fisher.test(matrix(c(0, cases-0, 6, controls-6), nrow=2, byrow=T))
The closest we get is when the E196X are 1 case and 5 controls: the Fisher test has it at an OR of 1.22 and a P-value of 0.60.
In total, then, the “FH Alzheimer disease dementia custom” phenotype is present in 53,734 / 380,992 (14%) of UK Biobank participants, and in 2/9 (22%) of those individuals with C-terminal PRNP PTVs, a small non-significant excess. It looks like there’s no association, at least for these 4 variants in the aggregate, or for E196X in particular.
inflammatory bowel disease cohort
A colleague was also able to share data from an inflammatory bowel disease (IBD) cohort included in gnomAD, which revealed that it contains 3 of the alleles of interest:
- Q186X in an IBD case
- Q223Rfs18X in an IBD case
- Q217X in a control (in fact, it’s in 2 controls in the IBD cohort though only 1 of them is present in gnomAD v4)
Thus, 2/4 of the alleles from IBD exomes are in cases. The cohort is approximately half cases and half controls, so this is the most likely outcome by chance. Still, the identity of the variants makes me speculate the ones seen in cases may not be coincidental. Q186X has also been seen in a prion disease case; while we have no information to confirm that it is highly penetrant, it certainly makes it more likely to be pathogenic. And Q223 frameshift is close to the known pathogenic cluster at 226-227. So in both cases, it seems plausible that the individuals with these variants are in gnomAD precisely because they were ascertained on IBD-like symptoms actually caused by a PRNP variant.
summary
In total, then, we tracked down data on 12 out of the 15 C-terminal PTV alleles in gnomAD that we were having trouble explaining. Here’s what we know now:
| codon | AC | variant | what we know |
|---|---|---|---|
| 155 | 1 | H155Lfs12X | UKBB FH Alzheimer case |
| 157 | 1 | Y157X | UKBB control |
| 159 | 1 | N159Qfs10X | UKBB control |
| 186 | 1 | Q186X | IBD case |
| 196 | 6 | E196X | UKBB, 1 FH Alzheimer case and 5 controls |
| 217 | 1 | Q217X | control in IBD cohort |
| 223 | 1 | Q223Rfs18X | IBD case |
| 226 | 1 | Y226X | unknown |
| 229 | 2 | G229X | unknown |
Putting together everything we know, below is my best guess as to how to classify the C-terminal PTVs that have ever been observed. I want to emphasize again that this is a best guess useful for thinking about the biology of the protein. We do not have the kind of confidence about many of these classifications that you would want to have in genetic counseling, and some of these could easily be overturned by new information (even from a single case) in the future, so take with a grain of salt:
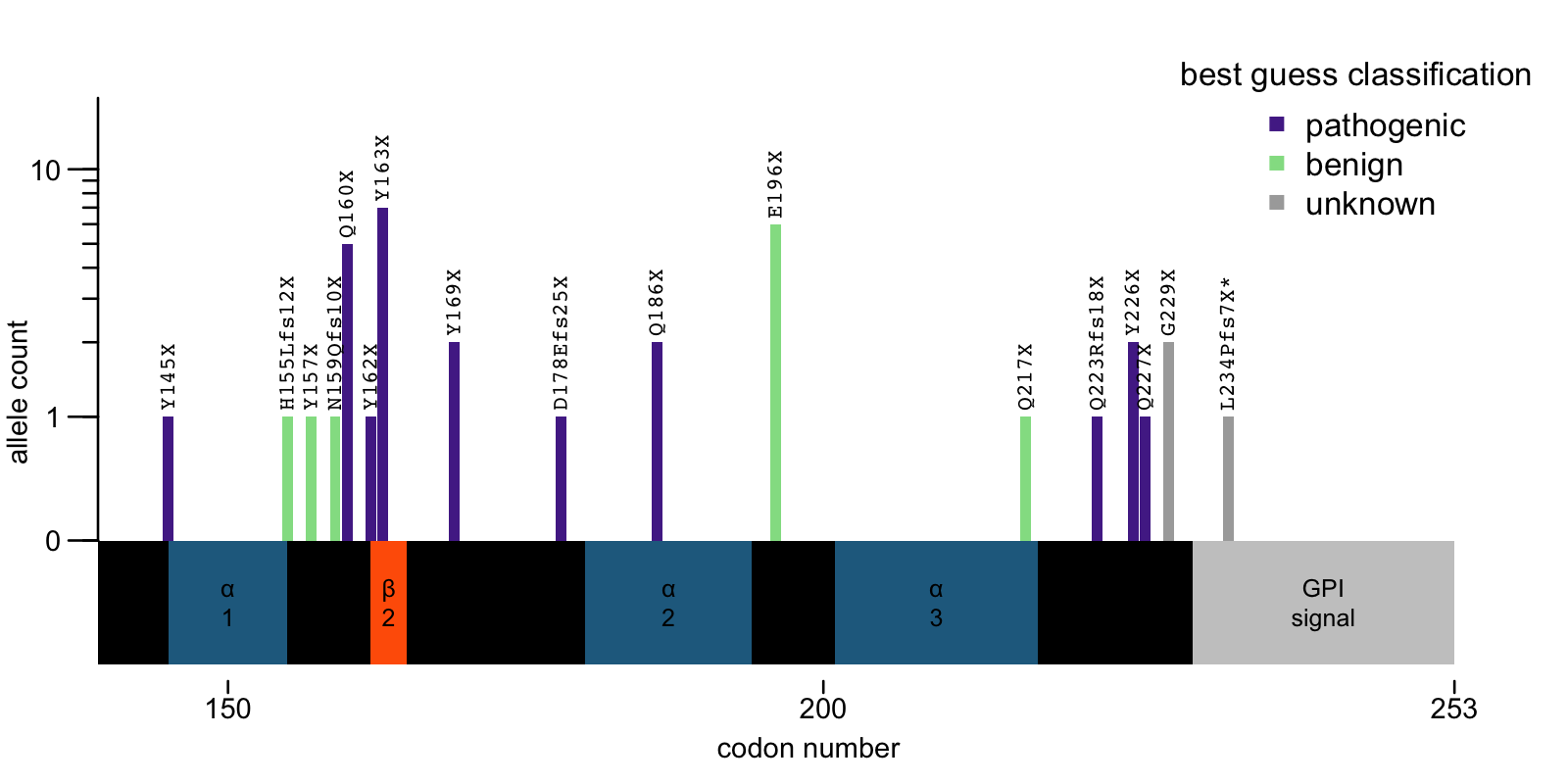
If true, then both hypotheses — phenotypic enrichemnt or position dependence — may be true to some degree. A handful of individuals with these variants in gnomAD, such as the IBD cases with variants at 186 and 223, may be in gnomAD because of their PRNP-related phenotypes. Most, however, have variants such as E196X or Q217X that appear to be benign, or at least of lower penetrance. Why, then, do some PTVs cause disease and others not?
You can appreciate that the putatively benign PTVs all occur at or near the end of alpha helices. These would cleanly shear off the end of the protein in a place that leaves a new C terminus ending in a piece of intact secondary structure, with little or no extra unfolded polypeptide hanging off the end. In contrast, the pathogenic variants occur in places that leave significant unstructured content occurring after the last bit of secondary structure. Could the presence of such unfolded polypeptide at the new C-terminus of the protein impact its propensity to misfold? Possibly. But the answer could easily be more complicated too — maybe we’ll one day learn that not all helices are created equal, and every single codon has its own penetrance. For now, there’s nothing we can do but wait for more data to accrue.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.