Mapping proteomics data to UniProt, RefSeq and gene symbols
update: this post was substantially revised on 2013-03-18 to correctly handle different isoforms of proteins
This week at work we finally got some new (human) proteomics data we’ve been waiting on for a while. It’s iTRAQ data, meaning that short subsequences of amino acids were quantified and then looked up in a reference proteome. I couldn’t find a good online introduction to how iTRAQ works, but the original paper introducing it appears to be Wiese 2007. In our case, we didn’t get any of the truly raw data, instead we got calls of protein levels for 2801 proteins across all our samples. Each protein is identified by its UniProt id, such as B4DFA2. To be precise, these are UniProtKB AC ids, distinct from UniProtKB ID ids.
A few weeks ago I had downloaded the UniProt human proteome from downloads > proteomes (see also the proteomes readme). At that time, UniProt had 87,621 proteins; now it has 87,656. According to the downloads page, UniProt updates every 4 weeks. Versioning is by date - see release notes. UniProt does have version numbers for proteins but not for releases – so as of March 6, 2013, protein A0AVT1 is on its version 70 while Q9UBC2 is on its version 111. Therefore the date, 2013-03-06, is the only proper version identifier for this release. Note that the proteome file itself contains no version identifiers at all – you’ll have to give it a name or put it in a folder that specifies the version. The proteome comes in FASTA format; my polyQ post has a very short python script to convert it to something tab-delimited (and parse the gene symbol, where present, out of the description) if you like.
In our proteomics dataset, each protein also has a “sequence” provided, which looks sort of, but not quite, like an amino acid sequence, for instance:
B4DFA2 sQLScVVVDDIER;lLDYVPIGPR;iAEESNFPFIk
If you grep for these all-caps substrings between the semicolons, they are present in the HUMAN.fasta proteome for the given UniProt id – just not in that order, and with plenty of other stuff between them. So these appear to be the individual fragments quantified in iTRAQ and then used to determine the identity of the protein. I’m not clear on how definitively these are able to distinguish the protein products of different splicing variants (isoforms) given that they don’t exhaustively cover the protein.
UniProt provides mapping tables so you can figure out what RefSeq transcripts these proteins correspond to. Under downloads > ID mapping data (see readme). The full mapping files are huge (~2GB) but I only want human, so HUMAN_9606_idmapping.dat.gz will do.
Three lines of bash:
wget ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/idmapping/by_organism/HUMAN_9606_idmapping.dat.gz gunzip HUMAN_9606_idmapping.dat.gz grep RefSeq_NT HUMAN_9606_idmapping.dat | cut -f1,3 > uniprot-refseqnt.tab
Will get you a two-column lookup table matching the UniProt ids to RefSeq transcripts:
$ head uniprot-refseqnt.tab P31946 NM_003404.3 P31946 NM_139323.2 P62258 NM_006761.4 Q04917 NM_003405.3 P61981 NM_012479.3 P31947 NM_006142.3 P27348 NM_006826.2 P63104 NM_001135699.1 P63104 NM_001135700.1 P63104 NM_001135701.1
Note it’s much shorter than the proteome:
$ cat uniprot-refseqnt.tab | wc -l 41614
And the relationship between proteins and transcripts is many-to-many. The number of unique proteins and transcripts represented is yet fewer:
$ cut -f1 uniprot-refseqnt.tab | sort | uniq | wc -l 25794 $ cut -f2 uniprot-refseqnt.tab | sort | uniq | wc -l 33654
Earlier I implied that the UniProtKB_AC ids are the unique identifiers of both my proteomics data and the UniProt reference human proteome. That is sort of true. In fact, both the reference proteome and my data contain different isoforms of the same proteins. For instance:
$ cat 2013-03-06/uniprot.tab | cut -f1 | grep Q9ULK2 Q9ULK2 Q9ULK2-2 Q9ULK2-3
And as far as UniProt seems to be concerned, only the first six characters Q9ULK2 comprise the UniProtKB_AC id, even though the longer form is required to uniquely specify an entry in the HUMAN.fasta reference, which I have here parsed into uniprot.tab. UniProt’s mapping files, including HUMAN_9606_idmapping.dat.gz which I have here parsed into uniprot-refseqnt.tab, as well as any mapping files you obtain through their web GUI (which we’ll do in a moment) only map the six character base IDs and will fail to match any isoforms with a suffix such as -2.
To prove to yourself that the RefSeq mapping table does not include isoforms, try getting the substring of the UniProtKB_AC id column starting from the seventh character:
cat 2013-03-06/uniprot-refseqnt.tab | awk '{print substr($1,7,10)}' | sort -u
# returns only the empty string
(Thanks to adambliss below for the sort -u trick)
Whereas if you do this for the reference proteome:
cat 2013-03-06/uniprot.tab | awk '{print substr($1,7,10)}' | sort -u
# you get everything from -2 to -35
So in a moment we will need to perform id mapping based on just the six-character core id, which I will hereafter call uniprotkb_ac6, and then handle isoforms separately. But since both my proteomics data and the reference proteome do contain isoforms, first I am interested to check whether all of my proteins are even in the reference. To do that, I cut from each file the column with just the full (isoform-specific) uniprot ids and use diff to compare the two and see which, if any, of my proteins are not in the reference proteome:
$ cat protein_Expression_matrices_all_2801_proteins.txt | cut -f1 | tail -n+9 | head -n-7 | sort -u > proteomics.uniprotkb_ac_full $ cat 2013-03-06/uniprot.tab | cut -f1 | sort -u > uniprot.uniprotkb_ac_full $ diff uniprot.uniprotkb_ac_full proteomics.uniprotkb_ac_full | grep $'>' | wc -l 223 $ diff uniprot.uniprotkb_ac_full proteomics.uniprotkb_ac_full | grep $'>' | head -3 > A2RRF3 > A6NE29 > A6NFM2
About 8% of my quantified proteins are missing from the reference. When I Googled the first example, A2RRF3, I found it was indeed a UniProt protein – but the reference symbol had managed to already change in the time since our proteomics provider generated the data a few weeks ago:
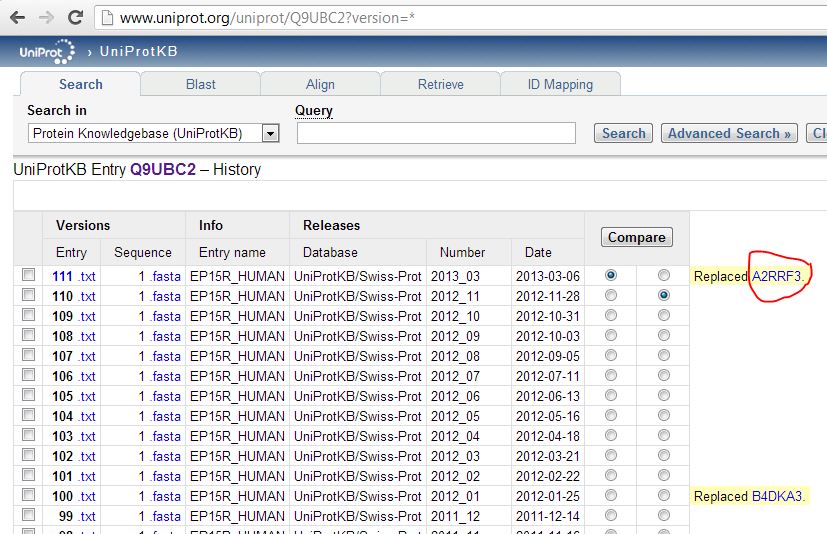
Still others had been wholesale deleted from UniProt:
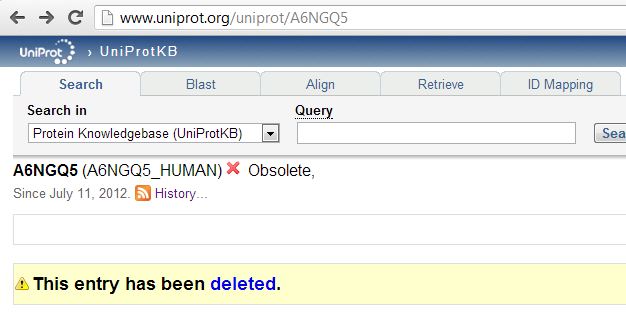
The UniProt FAQ on deletions says most deletions happen when the putative protein-coding sequence in the relevant DNA reference is deleted, or when someone realizes that an open reading frame doesn’t actually encode a protein like they thought it did. Clearly someone is wrong, then – either the deleted entries do actually have real proteins associated with them, or our proteomics provider is wrong to think they detected some of these proteins.
Now it’s clear we have three mapping issues to resolve:
- Old uniprotkb_ac6 ids to new uniprotkb_ac6 ids (*I learned the hard way this mapping cannot be done with the full id, only with the six character core)
- New uniprotkb_ac6 ids to RefSeq transcript ids
- Preserving isoform-specific information when doing mapping #1 above.
This means I need to separate the isoform-specific information from the uniprotkb_ac6 id but keep it around for later. I take our protein data file (which has old ids) and the reference (which has new 2013-03-06 ids) and add two columns to the front of each: the core uniprotkb_ac6, and then just the isoform suffix. For the proteomics data, I also start from line 9 and end at line -7 to remove the header and some other extraneous stuff:
cat protein_Expression_matrices_all_2801_proteins.txt | awk '{print substr($1,1,6)"\t"substr($1,7,10)"\t"$0}' | tail -n+9 | head -n-7 > proteomics.all
cat 2013-03-06/uniprot.tab | awk '{print substr($1,1,6)"\t"substr($1,7,10)"\t"$0}' > uniprot.all
(Aside: I am a useless use of cat enthusiast for (1) clarity and (2) ease of pressing up key and then editing the downstream commands without pressing left all the way to home. update – but crtl-a works!)
OK, so first time I did this I didn’t realize UniProt couldn’t map isoforms, so I uploaded a list of unique full ids including the isoform info (ex. Q92945-2) to UniProt’s Retrieve tool, and the isoform-specific ids were ‘rejected’:
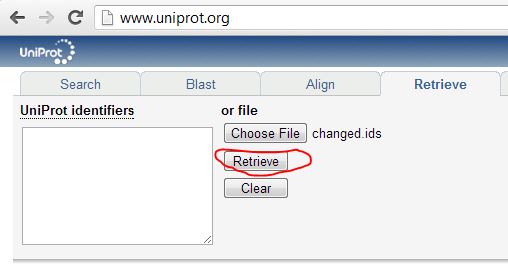
The output lists which proteins are rejected, replaced or deleted:
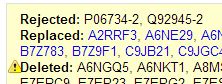
Clearly UniProt’s tool is designed to only handle the six-character ids. Here is how I get a list of just the unique (old) uniprotkb_ac6 ids from my proteomics data so that I can map those to their (new) counterparts in the current 2013-03-06 release:
$ cat proteomics.all | cut -f1 | sort -u > proteomics.uniprotkb_ac6_old $ wc -l proteomics.uniprotkb_ac6_old 2799 proteomics.uniprotkb_ac6_old
And when I upload that list to the Retrieve tool, none are ‘rejected’ – plenty are still ‘deleted’. Retrieve is useful for knowing what happened to your proteins but it doesn’t actually let you build mapping tables. The mapping between current uniprotkb_ac6 ids is not included in the HUMAN_9606_idmapping.dat.gz file we downloaded earlier, nor is it available through any API. As far as I can tell, this mapping is only possible through the web GUI’s ID Mapping interface:
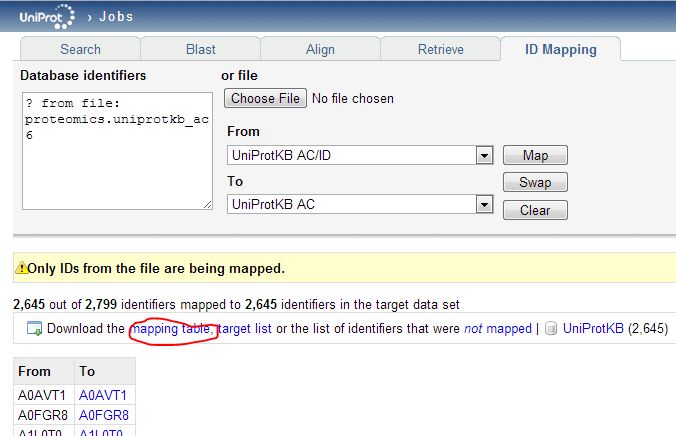
The mapping table you download will include all of the replaced ids, but the deleted ones will simply be omitted. You may notice that the “To” dropdown menu in the ID Mapping tool has “RefSeq Nucleotide” as one option. That looks easy: could we just map directly from the (outdated) UniProtKB_AC6 identifiers in the proteomics data to the current RefSeq transcript ids? Alternately could we download the list of new UniProtKB_AC6 ids, then re-upload those to get RefSeq transcripts? You’d think so, but when I try either of these approaches, very few proteins map (only 877 of 2646 unique new UniProtKB_AC6 ids find a corresponding transcript) and some things fail to map that obviously should – for instance, Q9UBC2 does not map to a transcript:
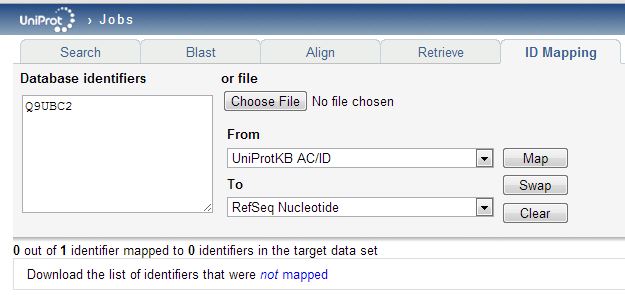
Even though it totally does have RefSeq transcripts associated with it:
![]()
So to my mind the safest thing to do is use UniProt’s ID Mapping service to convert old to new UniProtKB_AC6 ids, then just look those up in the full human mapping table we downloaded and grepped earlier (uniprot-refseqnt.tab). RefSeq mappings (as shown above for instance) are present in that table:
$ grep Q9UBC2 uniprot-refseqnt.tab Q9UBC2 NM_001258374.1 Q9UBC2 NM_021235.2
At this point I’ve got four files:
- Proteomics data, i.e. protein levels
- Mapping from old to new UniProt ids
- Mapping from new UniProt ids to RefSeq transcripts
- UniProt human proteome
And here’s the table schema if I want to join these in SQL:
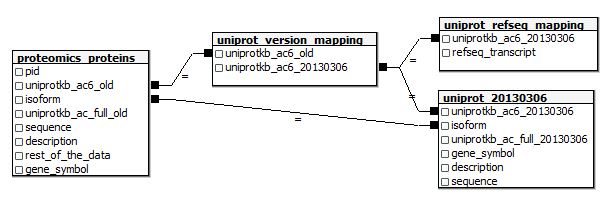
Note that I’m making the assumption that even if a protein’s uniprotkb_ac6 id has changed, the old protein’s -2 isoform will still correspond to the new protein’s -2 isoform. I don’t know whether that’s a safe assumption (and nothing on UniProt’s website appears to answer this question) so I’ll need to be careful about isoform-specific analyses later on. But if you dig the above E-R diagram anyway, here are the create table statements:
drop table if exists proteomics_proteins;
create table proteomics_proteins (
pid serial primary key,
uniprotkb_ac6_old varchar,
isoform varchar,
uniprotkb_ac_full_old varchar,
sequence varchar,
description varchar,
rest_of_the_data varchar
);
drop table if exists uniprot_version_mapping;
create table uniprot_version_mapping (
uniprotkb_ac6_old varchar,
uniprotkb_ac6_20130306 varchar
);
drop table if exists uniprot_20130306;
create table uniprot_20130306 (
uniprotkb_ac6_20130306 varchar,
isoform varchar,
uniprotkb_ac_full_20130306 varchar,
gene_symbol varchar,
description varchar,
sequence varchar
);
drop table if exists uniprot_refseq_mapping;
create table uniprot_refseq_mapping (
uniprotkb_ac6_20130306 varchar,
refseq_transcript varchar
);
-- copy the tables from disk here
alter table proteomics_proteins add column gene_symbol varchar;
update proteomics_proteins set gene_symbol = substring(substring(description from 'GN=[A-Za-z0-9]*') from 4);
Note that more than one protein can match one gene symbol, so purists might spin that off into a separate match table – but since the gene symbols are already included in the reference proteome and in my proteomics data, I just left them there. When I join my new tables together:
select distinct pp.uniprotkb_ac_full_old
from proteomics_proteins pp, uniprot_version_mapping uv, uniprot_refseq_mapping ur, uniprot_20130306 u
where uv.uniprotkb_ac6_20130306 = ur.uniprotkb_ac6_20130306
and uv.uniprotkb_ac6_20130306 = u.uniprotkb_ac6_20130306
and pp.uniprotkb_ac6_old = uv.uniprotkb_ac6_old
and pp.isoform = u.isoform
;
I get 2160 of my 2801 proteins. This set is limited to those that map to both (1) at least one RefSeq transcript and (2) a corresponding isoform in the current UniProt reference. This set includes Q9UBC2, the example I gave above that didn’t map to RefSeq through the ID Mapping tool online but does map using the mapping table I created locally. So 2160 of 2801 is an improvement, but still, what’s with some proteins not mapping to a transcript? When I look at the proteins that still don’t map:
select *
from uniprot_version_mapping uv, uniprot_20130306 u
where uv.uniprotkb_ac6_20130306 = u.uniprotkb_ac6_20130306
and not exists (select null from uniprot_refseq_mapping ur where ur.uniprotkb_ac6_20130306 = uv.uniprotkb_ac6_20130306)
;
I find that they almost all do have gene symbols in the UniProt reference proteome (here’s a quick sample):
![]()
So it isn’t as though no one knows the genomic location that codes for these proteins. Apparently they just haven’t been assigned to a specific location yet. In the downstream analysis, then, it’s not yet clear whether it will make sense to use protein, transcript or gene as the relevant unit of analysis. Gene would be most inclusive, particularly since even many of the ‘deleted’ or ‘rejected’ UniProt ids from my proteomics data still have gene symbols. But gene symbols are neither universal nor unique among my quantified proteins:
select *
from proteomics_proteins pp
where gene_symbol is null
;
-- 6 proteins have no gene_symbol
select gene_symbol, count(*) n
from proteomics_proteins pp
group by 1
order by 2 desc
;
-- some gene_symbol are associated with > 1 protein
The multiple proteins per gene symbol includes 20 different proteins that all have the gene symbol listed simply as ‘HLA’. Even once mapping to transcripts and genes is done, the question of how to handle ambiguity and multiplicity in the downstream analysis will not be trivial.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.